Hoffnung für junge Patienten mit der Charcot-Marie-Tooth-Erkrankung
Forscher können an Ratten durch frühzeitige Therapie mit dem Kombinationspräparat PXT3003 den Krankheitsbeginn verzögern und Symptome mildern
Die Charcot-Marie-Tooth-Erkrankung Typ 1A (CMT1A) ist die häufigste erbliche Erkrankung des peripheren Nervensystems beim Menschen. Patienten leiden oft schon im Kindesalter an Muskelschwäche, die sich mit zunehmendem Alter verschlimmert. Grund dafür ist eine fehlerhafte Entwicklung der Schwannzellen, wodurch die Reizweiterleitung über die Nervenzellen an die Muskeln beeinträchtigt wird. Forscher am Max-Planck-Institut für experimentelle Medizin und der Universitätsmedizin in Göttingen haben gezeigt, dass Ratten von einer frühzeitigen Behandlung während der kritischen Phase der Schwannzell-Entwicklung mit einer Kombination aus niedrig dosierten Medikamenten langfristig profitieren. Mithilfe der sogenannten PXT3003-Therapie wird das Auftreten von Symptomen teilweise verhindert, und der Beginn der Krankheit verschiebt sich bis ins Erwachsenenalter. Das gibt die Hoffnung, durch eine frühzeitige PXT3003-Behandlung von Kindern und Jugendlichen mit CMT1A den weiteren Verlauf der Krankheit entscheidend zu beeinflussen.
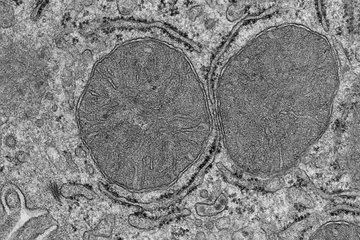
, einer an Charcot-Marie-Tooth (CMT1A) erkrankten (Mitte) und einer mit PXT3003 behandelten CMT1A-Ratte (rechts). Die Behandlung bewirkt eine verbesserte Myelinisierung im CMT1A Rattenmodell, die gesunden Tieren nahe kommt.")

Patienten mit der Charcot-Marie-Tooth Erkrankung 1A tragen auf dem Chromosom 17 eine zusätzliche Kopie des Gens Pmp22. Durch diesen Gendefekt entwickeln die Betroffenen eine langsam fortschreitende Schädigung der Nervenzellen, weshalb bereits im Kindesalter erste Symptome wie Gehschwierigkeiten oder Deformationen der Füße auftreten können. Die Muskeln können durch die geschädigten Nerven die Befehle des Gehirns nur unzureichend empfangen. Dies führt mit der Zeit zum Abbau der Muskeln, der sich vor allem in schwindender Kraft in Armen und Beinen zeigt. Im weiteren Verlauf kommt es zu Sensibilitätsstörungen wie Taubheit, Kribbeln oder Schmerzen in den Extremitäten. Bisher gibt es keine zugelassene Therapie, die die Ursachen der CMT1A-Erkrankung bekämpft.
Genetisch veränderte Ratten mit Krankheitssymptomen
Nervenzellen besitzen lange Fortsätze, die Axone, die für die Weiterleitung der Nervenimpulse verantwortlich sind. Schwannzellen umhüllen die Axone mit einer isolierenden Schicht aus Myelin und stellen somit die schnelle und effiziente Weiterleitung der elektrischen Nervenimpulse sicher. Bei der Charcot-Marie-Tooth-Erkrankung 1A wird durch die zusätzliche Kopie des Gens Pmp22 das Periphere Myelin Protein 22 (PMP22) überproduziert. Um die molekularen und zellulären Ursachen besser zu verstehen, untersucht Michael Sereda vom Max-Planck-Institut für experimentelle Medizin und der Universitätsklinik in Göttingen mit seinen Kollegen genetisch veränderte Ratten. Diese Tiere tragen ebenfalls eine zusätzliche Kopie des Gens Pmp22 und entwickeln ähnliche Symptome wie der Mensch. Die Experimente haben ergeben, dass die Schwannzellen in diesen Tieren durch die Überaktivität des Gens Pmp22 nicht richtig ausreifen können und es deshalb zu einer gestörten Funktion der Nervenfasern kommt.
Da die Erkrankung schon früh erkennbar ist und anfangs meist mit milden Symptomen einhergeht, könnte eine frühzeitige, effektive Therapie für junge CMT1A-Patienten von großem Nutzen sein. Kinder betroffener Eltern können sogar vor Auftreten erster Symptome genetisch untersucht werden. Thomas Prukop, Michael Sereda und ihre Kollegen haben daher untersucht, welchen Effekt es auf den Krankheitsverlauf hat, wenn die Entwicklung der Schwannzellen in den genetisch veränderten Ratten frühzeitig beeinflusst wird. Dazu verabreichten sie jungen Tieren vom sechsten bis zum 18. Lebenstag PXT3003, eine Kombination aus niedrig dosierten Medikamenten, die jeweils überschüssige Abschriften des Pmp22-Gens reduzieren sollten.
Therapie verbessert motorische Fähigkeiten
„Durch die kurze Therapie mit PXT3003 innerhalb der ersten zwei Lebenswochen verbesserten sich einige motorischen Fähigkeiten der Ratten so sehr, dass sie mit denen gesunder Artgenossen langfristig vergleichbar waren“, sagt Thomas Prukop. Der Krankheitsbeginn verzögert sich bei den behandelten Tieren dadurch bis ins Erwachsenenalter, auch wenn nicht alle Auffälligkeiten von der kurzfristigen Therapie völlig verschwanden. Auf molekularer Ebene sahen die Forscher, dass PXT3003 die überschüssigen Abschriften des Pmp22-Gens verringert und damit die Signalwege unterstützt, die für die Ausreifung der Schwannzellen verantwortlich sind. Die bessere Entwicklung der Schwannzellen könnte ein Grund sein, dass die Versorgung der Axone und die Reizübertragung an die Muskulatur langfristig unterstützt wurde.
PXT3003 gilt als sichere Therapie, die bereits in einer klinischen Studie an erwachsenen CMT1A-Patienten als wirksam getestet wurde. „Aufgrund unserer Ergebnisse hoffen wir, dass der frühzeitige Behandlungsbeginn mit PXT3003 eine Therapieoption für Kinder und Jugendliche mit CMT1A darstellen könnte“, erklärt Michael Sereda. Eine klinische Studie, bei der erstmals betroffene Kinder mit PXT3003 behandelt werden sollen, ist bereits für dieses Jahr geplant.
MN/HR