Der Anfang vom Ende
Max-Planck-Forschende zeigen, dass bei der Ort des Transkriptionsbeginns meist auch den Ort des Transkriptionsendes bestimmt
Jedes Gen in unserer DNA hat einen Anfang und ein Ende. Die richtige Zuordnung der Anfangs- und Endpunkte eines Gens ist entscheidend für die Herstellung funktionsfähiger Eiweiße. Viele Forschungsarbeiten beschäftigten sich mit der Frage, was bestimmt, wann und an welcher Stelle der DNA ein Gen „beginnt“. Doch wo ein Gen „endet“, ist eine andere, eher vernachlässigte Forschungsfrage: Es wurde stets angenommen, dass die Auswahl der Enden von nachgeschalteten Elementen und äußeren Faktoren abhängt. In ihrer neuesten Studie hat ein Forschungsteam des Max-Planck-Instituts für Immunbiologie und Epigenetik jedoch die erstaunliche Entdeckung gemacht, dass bei den meisten unserer Gene der Ort des Transkriptionsbeginns auch den Ort des Transkriptionsendes bestimmt. Dieses Phänomen ist artenübergreifend und spielt eine entscheidende Rolle für die Identität und Funktionalität von Zellen.
 und zwei mögliche Endstellen (TES) enthält. Die schwarzen Kästen im Genmodell zeigen Sequenzen, die in Eiweiß übersetzt werden. Bei der herkömmlichen mRNA-Sequenzierung mit kurzen Leseabschnitten, bei der das Signal die Anhäufung von Leseabschnitten darstellt, ist es nicht möglich, verschiedene Moleküle zu unterscheiden. Bei der Long-Read-Sequenzierung stellt jede horizontale Linie ein mRNA-Molekül vom Anfang bis zum Ende dar. Für das Gen stathmin können wir sehen, dass TSS1 das Ende an Position TES1 vorgibt und TSS2 zu TES2 führt.")
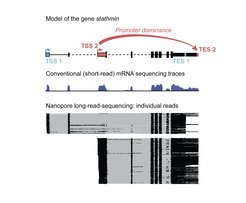
Alle Zellen unseres Körpers enthalten die gleiche DNA-Sequenz. Jedoch wird die eigentliche Identität einer Zelle und deren Funktionen davon bestimmt, welche Gene an einem bestimmten Ort und zu einem bestimmten Zeitpunkt aktiv sind. Diese aktiven Gene werden von der DNA-Vorlage in einzelne Boten-RNA-Moleküle (mRNA) übertragen und kodieren die Eiweiße, die die Zelle benötigt.
Dabei bindet an bestimmten Stellen auf der DNA, den so genannten Promotern, eine komplexe molekulare Maschinerie und beginnt mit der Transkription von DNA-Sequenzen in mRNA. Interessanterweise gibt es bei den meisten Genen mehrere Stellen, an denen die Transkription beginnen und auch enden kann. Das bedeutet, dass die mRNAs für jedes Gen je nach Start- oder Endstelle unterschiedlich sein können. Die Expression eines Gens in verschiedenen Varianten erweitert die Vielfalt und Funktionalität des Genoms um ein Vielfaches. Gleichzeitig wird die Erforschung der Gene dadurch auch wesentlich komplexer.
Boten-RNA von Anfang bis Ende auslesen
Ein Forschungsteam am Freiburger Max-Planck-Institut für Immunbiologie und Epigenetik ist der Frage nachgegangen, wie viele verschiedene Start- und Endstellen jedes Gen verwendet, welcher Kombination vorliegen und ob diese Kombinationen unter verschiedenen Bedingungen unterschiedlich sind. „Die Beantwortung dieser Frage stellte uns zunächst vor große technische Hürden, da wir jedes einzelne mRNA-Molekül von allen aktiven Genen vom Anfang bis zum Ende auslesen mussten. Das ist eine gigantische Aufgabe, die so bisher noch nicht in Angriff genommen wurde“, sagt Valérie Hilgers, Leiterin der Forschungsgruppe am Max-Planck-Institut für Immunbiologie und Epigenetik und Mitglied des Exzellenzclusters CIBSS – Centre for Integrative Biological Signalling Studies an der Universität Freiburg.
Das Team nutzte eine eigens für die Forschungsfrage optimierte Sequenzierungstechnologie, um die einzelnen mRNAs komplett auszulesen. Bei der herkömmlichen Short-Read-Sequenzierung wird jede mRNA in kürzere Fragmente zerlegt, die anschließend durch Kopieren verstärkt und dann sequenziert werden, um den „Read“ zu erzeugen. Mit Hilfe bioinformatischer Techniken werden die Read-Fragmente dann wie ein Puzzle zu einer kontinuierlichen Sequenz zusammengesetzt.
Um mRNA-Informationen in voller Länge über das gesamte Genom in verschiedenen Geweben der Fruchtfliege Drosophila einschließlich des Fliegenhirns zu erhalten, hat sich das Labor von Valérie Hilgers mit der Forschungseinheit für Tiefensequenzierung des Max-Planck-Instituts für Immunbiologie und Epigenetik zusammengetan, um sogenannte Long-Read-Sequenzierungstechnologien für ihre Bedarfe anzupassen. „Die Long-Read-Sequenzierung ermöglicht die Erfassung wesentlich längerer Sequenzierungsabschnitte als die Standard-Sequenzierung. Allerdings mussten wir für unsere Zwecke diese Technologie sogar noch weiter optimieren und die typische Leselänge um ein Vielfaches erhöhen, um mRNA-Informationen in voller Länge in unseren verschiedenen Modellsystemen zu erhalten,“ sagt Carlos Alfonso-Gonzalez, der Erstautor der Studie. Neben der Fruchtfliege Drosophila bezog das Team auch ein menschliches Modell des Nervensystems in die Studie ein und sequenzierten sogenannte zerebrale Organoide. Dabei handelt es sich um „Mini-Gehirne", also dreidimensionales Hirngewebe, das aus induzierten pluripotenten Stammzellen in Kultur gezüchtet wurde.
Endpunkte der Transkription stehen schon zu Beginn fest
Die gewonnenen Daten geben einen bislang beispiellosen Einblick in die Transkription einzelner Gene. „Wir haben festgestellt, dass die Startstellen der Transkription und die Endstellen nicht zufällig miteinander kombiniert werden. Vielmehr sind die Startstellen oft spezifisch mit bestimmten Endstellen verbunden“, erklärt Valérie Hilgers. Diese Verknüpfung ist kausal: In den Eierstöcken, zum Beispiel, setzt die künstliche Aktivierung einer Startstelle, die normalerweise nur im Gehirn verwendet wird, die normale Endstellen außer Kraft und veranlasst künstlich die Verwendung der Endstellen wie in Gehirnzellen. Dies zeigt die entscheidende Rolle der Startstellen für die jeweilige RNA-Landschaft, die für jeden Gewebetyp einzigartig ist.
Ein Phänomen stach jedoch besonders hervor. „Bestimmte Startstellen zeigen ein überraschendes Dominanzverhalten. Sie setzen sich über konventionelle Signale zur Beendigung der Transkription hinweg, verdrängen andere Startstellen und bewirken die Selektion von bestimmten Endstellen. Daher haben wir sie »dominant promoters« genannt“, sagt Carlos Alfonso-Gonzalez. Darüber hinaus stellte das Team fest, dass die Interaktionen zwischen diesen dominanten Promotern und den mit ihnen verbundenen Endstellen durch unterschiedliche epigenetische Signaturen gesteuert werden. Wichtig ist, dass die Ergebnisse in Drosophila-Gehirnzellen auch in den menschlichen Hirnorganoiden reproduziert werden konnten. Dies zeigt, dass es sich um einen hochkonservierten, vielleicht universellen, Mechanismus handelt, der die Produktion funktioneller Eiweiße reguliert und entscheidend für die Funktionalität der Zelle ist.
Doch welche biologische Bedeutung könnte dieser bislang unbekannte Mechanismus haben? Durch eine eingehende Analyse der Sequenzierungsdaten von Dutzenden verschiedener Spezies entdeckten die Freiburger Forscherinnen und Forscher, dass Start- und Endstellen eine Koevolution durchlaufen haben: Über Millionen von Jahren Evolution zwischen den Arten wurden einzelne Nukleotidänderungen am Genanfang an „dominant promoters“ von Änderungen am entsprechenden Genende begleitet. „Wir interpretieren diese Beobachtung als eine Art Stoß durch die Evolution, um die Interaktion zwischen den beiden Enden des Gens aufrechtzuerhalten, was auf eine erhebliche Bedeutung dieser Kopplungen für die Fitness der Tiere schließen lässt“, erläutert Valérie Hilgers.
VH/MR
Teaserbild: Eine künstlerische Darstellung verschiedener Startstellen, die Türen zu völlig unterschiedlichen Welten öffnen. Bild generiert vom MPI-IE PR-Abteilung durch KI-Generierung mithilfe von midjourney // Creative Commons Noncommercial 4.0 Attribution International











