Forschungsbericht 2014 - Max-Planck-Institut für medizinische Forschung
Wege zur effizienten Synthese von neuen Glykopeptid-Antibiotika
Max-Planck-Institut für medizinische Forschung, Heidelberg
Abteilung Biomolekulare Mechanismen
Antibiotika sind einer der größten Erfolge in der Geschichte der Medizin und haben die Lebenserwartung der Menschen dramatisch erhöht [1]. Die ersten Antibiotika waren Naturstoffe, mit denen Bakterien oder Schimmelpilze versuchen, das Wachstum von konkurrierenden Mikroorganismen zu hemmen. Bereits bis zur Mitte des 20. Jahrhunderts wurden zahlreiche Antibiotika entwickelt und erfolgreich angewandt; seither wurden viele weitere, teilweise viel nützlichere, durch künstliche Änderungen geschaffen. Allerdings verlangsamte sich in den letzten Jahrzehnten die Entwicklung neuer Antibiotika stark, sodass die zunehmende bakterielle Resistenz heute eine ernsthafte neue Bedrohung darstellt. Um die künftige klinische Verwendung von Antibiotika zu sichern, müssen neue Antibiotikaklassen entwickelt und die bestehenden Klassen modifiziert werden, um ihre Wirksamkeit trotz steigender Zahl resistenter Keime aufrechtzuerhalten.
Seit dem Ende der 1950er Jahre gewannen Glykopeptid-Antibiotika, wie Vancomycin und Teicoplanin, große Bedeutung als letztes Mittel im Kampf gegen grampositive Bakterien [1]. Diese komplexen Verbindungen bestehen in ihrer molekularen Struktur aus einer Peptidkette mit zahlreichen chemischen "Dekorationen". Die dreidimensionale Form der Kette wird durch ein Muster von Querverbindungen zwischen aromatischen Aminosäuren innerhalb der Kette bestimmt und ist für die antibakterielle Wirkung essenziell. Diese hängt davon ab, dass das Molekül einen bestimmten Baustein bei der Synthese der bakteriellen Zellwand blockiert und somit die Bakterien abtötet. In dieser Hinsicht sind Glykopeptid-Antibiotika ungewöhnlich, denn die meisten anderen Antibiotika stören den bakteriellen Stoffwechsel. Dieser ungewöhnliche Mechanismus bietet einen Vorteil: Resistenzen gegenüber den Glykopeptid-Antibiotika entstehen zwar, doch geschieht dies langsamer als bei anderen Antibiotikaklassen.
Bestehende und neue Ansätze zur Herstellung von Glykopeptid-Antibiotika
Wegen ihrer komplexen Struktur lassen sich die Glykopeptid-Antibiotika schwer im Labor synthetisieren. Vor allem die voll-chemische Herstellung im industriellen Maßstab ist nicht praktikabel. Daher gehen alle klinisch wichtigen Glykopeptid-Antibiotika (natürlich vorkommende wie auch modifizierte Formen) letztendlich auf Fermentation zurück [1; 2], wie sie auch für viele andere Pharmazeutika üblich ist. Bei der Fermentation werden Naturstoffe aus Kulturen der natürlich produzierenden Organismen geerntet und weiterverarbeitet. Bei der neuen Generation der Glykopeptid-Antibiotika wird ein durch Fermentation produziertes Naturprodukt verwendet und dann die Oberfläche des Moleküls künstlich modifiziert. Dadurch kann der Wirkungsmechanismus beibehalten, die Effizienz jedoch erhöht werden. Besonderer Vorteil dieser Strategie ist, dass der produzierende Organismus niemals mit dem modifizierten Antibiotikum in Berührung kommt und daher auch keine Resistenz dagegen entwickeln muss, die wiederum auf andere, potenziell pathogene Bakterien übertragen werden könnte. Gegen bereits resistente Keime sind diese Derivate jedoch oftmals nicht wirksam, da weitreichendere Veränderungen des Grundgerüsts notwendig wären. Ein schönes Beispiel für ein solch wirksames Derivat ist ein jüngst synthetisierter Vancomycin-Ersatzstoff, der Resistenzen gegen herkömmliches Vancomycin überwinden kann [3]. Wie bereits erklärt, kann diese Form allerdings nicht in klinisch relevanten Mengen hergestellt werden, weil es nicht von einem natürlichen Organismus produziert wird. Für die effiziente Herstellung modifizierter Glykopeptid-Antibiotika in ausreichenden Mengen wäre ein besseres Verständnis der zugrunde liegenden Biosynthese erforderlich, das man sich dann in einem halb-synthetischen Ansatz zunutze machen könnte. Forscher am MPI für medizinische Forschung berichten nun über einen vielversprechenden Ansatz für die Herstellung neuer Glykopeptid-Antibiotika.
Biosynthese der Glykopeptid-Antibiotika
Die Biosynthese der Glykopeptid-Antibiotika wird seit vielen Jahren erforscht [2]. Sie erfolgt in drei Schritten. Im ersten Schritt (A) werden die Bausteine hergestellt: dies sind Aminosäuren, unter denen auch solche sein können, die in normalen Proteinen nicht vorkommen. Im zweiten Schritt (B) werden diese Bausteine zu einer Peptidkette zusammengefügt. Dies geschieht nicht mittels Ribosomen, wie bei normalen Proteinen, sondern über ein molekulares „Fließband“, bei dem aufeinander folgende Funktionsmodule das Peptidantibiotikum Aminosäure für Aminosäure aufbauen (Abb. 1).

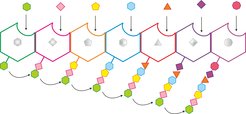
Abb. 1: Biosynthese der Glykopeptid-Antibiotika. Im produzierenden Bakterium werden im 2. Schritt Glykopeptid-Antibiotika aus ihren Aminosäuren-Bausteinen zusammengestellt: nicht an Ribosomen, sondern an speziellen „Fließbändern“.
Dabei wird die Abfolge der Aminosäuren in der Peptidkette von den Proteindomänen der einzelnen Module bestimmt und ist nicht von DNA oder RNA kodiert. Das wiederum ermöglicht die Verwendung von ungewöhnlichen Aminosäuren und erlaubt es, strukturell vielfältige Peptide zu produzieren. Im letzten Schritt (C) wird die Peptidkette durch Querverbindungen ergänzt, um die erforderliche 3-D-Struktur zu stabilisieren, die dann anschließend weiter modifiziert werden kann [4].
Aus Sicht der hier beschriebenen Fragestellung ist Schritt C der interessanteste Schritt der Biosynthese, weil eben diese Querverbindungen maßgeblich für die antibiotische Funktion und gleichzeitig das Haupthindernis bei der Synthese im Labor sind, da sie nur über viele Umwege erreicht werden. Daher wäre es wichtig, besser zu verstehen, wie dieser Schritt in der Natur erfolgt. Das Querverbinden wird in vivo durch oxidierende Enzyme, die Cytochrome P450, katalysiert [4]. Enzyme dieser Klasse sind an vielen zellulären Reaktionen beteiligt [4; 5]. Im Fall der Glykopeptid-Antibiotika haben sie die Aufgabe, verschiedene Querverbindungen (Abb. 2) zwischen aromatischen Seitenketten des Peptids in einer bestimmten Reihenfolge herzustellen [4; 5].

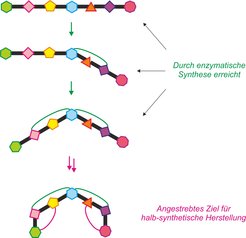
Um diese Mechanismen jedoch genauer zu studieren, mussten In-vitro-Experimente an Cytochromen P450 durchgeführt werden. Von früheren Studien ist bereits bekannt, dass die Querbindungen bereits dann entstehen, wenn das Peptid noch an das „Fließband“ gebunden ist [6]. Abgesehen von der ersten Querverbindung konnten jedoch bis vor kurzem keine Enzymaktivitäten gemessen werden [6; 7]. Damit war klar, dass unser Verständnis dieses Prozesses lückenhaft ist. Detaillierte Kenntnisse über diesen Mechanismus würden die Wissenschaftler jedoch in die Lage versetzen, den natürlichen Prozess mit den Möglichkeiten der synthetischen Chemie zu kombinieren, um neue Glykopeptid-Antibiotika effizient herzustellen und zu testen.
Forschungsfortschritte
Aktuelle Arbeiten am MPI für medizinische Forschung werfen ein neues Licht auf dieses Problem: Sie zeigen, dass für das Andocken weiterer Cytochrom-P450-Enzyme an das „Fließband“ eine bestimmte Domäne notwendig ist, deren Funktion bisher unbekannt war [8]. Diese sogenannte „X-Domäne” ähnelt anderen Domänen, die für die Herstellung von Peptidbindungen typisch sind. Aufgrund des molekularen Aufbaus des „Fließbands“ war es jedoch offensichtlich, dass dies nicht die Funktion der X-Domäne sein konnte. Die Strukturaufklärung bestätigte schließlich, dass die für die Peptidbindungsherstellung [8] verantwortlichen Bereiche in dieser Domäne deutlich verändert waren. Somit musste diese in allen Systemen auftretende und stark konservierte X-Domäne eine andere Aufgabe haben. Anhand verschiedener biochemischer Methoden wurde gezeigt, dass die X-Domäne mit den querverbindenden Cytochrom P450-Enzymen in Wechselwirkung tritt [8]. Um dies genauer zu prüfen, wurde die Struktur eines Komplexes des ersten P450-Enzyms (OxyB) mit der X-Domäne kristallographisch bestimmt (Abb. 3).
 im Kontaktbereich zwischen der X-Domäne und dem querverbindenden OxyB-Enzym.")
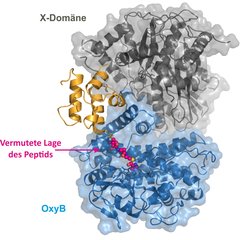
Dies zeigte, dass die Anordnung der Proteine ideal ist für die Vernetzungs-Reaktion, bei der das Peptid von der Domäne, die im Fließband direkt neben der X-Domäne liegt, dem P450-Enzym dargeboten wird [8].
Nachdem bewiesen war, dass die X-Domäne mit den querverbindenden Enzymen interagiert, musste die Rolle der X-Domäne bei der Aktivität der Cytochrom-P450-Enzyme genauer geklärt werden. In Abwesenheit der X-Domäne konnten keine enzymatischen Aktivitäten über die erste Querverbindung hinaus beobachtet werden, und es galt zu testen, ob ihre Anwesenheit etwas daran ändern würde. Um derartige Experimente durchzuführen, ist es zwingend notwendig, die erforderlichen Peptidsubstrate zur Verfügung zu haben. Daher wurde erst kürzlich am MPI für medizinische Forschung eine schnelle und effiziente Synthese entwickelt [9; 10], die es möglich machte, eine Vielzahl an Experimenten mit verschiedenen Peptiden durchzuführen. Mit Hilfe dieser Peptidsubstrate stellte sich heraus, dass die Peptide für mehrere Glykopeptid-Antibiotika (darunter auch das klinisch relevante Teicoplanin) nur dann querverbunden werden, wenn die X-Domäne anwesend ist [8]. Also benötigt auch der erste Vernetzungsschritt die X-Domäne. Das heißt auch, dass das inzwischen gut erforschte Vancomycin-System in der Hinsicht ungewöhnlich ist, da hier die X-Domäne nicht für die erste Querverbindung benötigt wird. Noch wichtiger war die Erkenntnis, dass die ersten und zweiten Enzyme der Teicoplanin-Biosynthese zusammen sowohl die erste als auch die zweite Querverbindung im Peptid herstellen können – dies konnte zum ersten Mal in vitro beobachtet werden [8]. Des Weiteren behindern Mutationen, die in der X-Domäne die Wechselwirkung mit den oxidierenden P450-Enzymen verhindern, auch deren Aktivität, was die Bedeutung dieser Wechselwirkung weiter unterstreicht [8].
Aussichten
Die neuen Erkenntnisse über die Biosynthese der Glykopeptid-Antibiotika bringen die vollständige Nachahmung der Quervernetzung von Peptiden im Reagenzglas in greifbare Nähe. Dies würde in Kombination mit der effizienten Synthese neuer Peptidsubstrate ein In-vitro-System zur Herstellung modifizierter Glykopeptid-Antibiotika ermöglichen, mit dem auf den rasanten Anstieg der Antibiotika-Resistenzen bei pathogenen Bakterien reagiert werden könnte.
Literaturhinweise
Journal of Antibiotics 67, 631-644 (2014)
doi:10.1038/ja.2014.111
Journal of Antibiotics 67, 31-41 (2014)
doi:10.1038/ja.2013.117
Journal of the American Chemical Society 134, 1284-1297 (2011)
Angewandte Chemie, International Edition 43, 6709-6713 (2004)
ChemBioChem, 15, 2719-2728 (2014)
doi:10.1002/cbic.201402441
Nature 2015 Feb 9 (epub ahead of print)
doi: 10.1038/nature14141
Organic and Biomolecular Chemistry 13(7), 2012-21 (2015)
doi: 10.1039/C4oB02452D
Organic Letters 16, 2454-2457 (2014)
doi: 10.1021/ol500840f