Forschungsbericht 2013 - Max-Planck-Institut für Biophysik
Aktuelle Forschung aus der Strukturbiologie
Arbeitsgruppe Werner Kühlbrandt
Die Gruppe konzentriert sie sich auf zwei Forschungsrichtungen: 1. Struktur und molekulare Mechanismen von Membranproteinen mittels Elektronen- und Röntgenkristallographie und 2. funktionale Anordnung großer Proteinkomplexe mittels Einzelpartikel- und Kryo-Elektronenmikroskopie sowie Kryo-Elektronentomographie (Kryo-ET).
1. Untersuchung der Struktur und Funktion von Membranproteinen
Channelrhodopsin-2 (ChR2) ist ein Kationen-selektiver, lichtgesteuerter Ionenkanal aus der Grünalge Chlamydomonas reinhardtii, der sich zu einem vielseitigen Objekt und Instrument in der Optogenetik entwickelt hat. In Zusammenarbeit mit Ernst Bamberg und Christian Bamann aus der Abteilung Biophysikalische Chemie des Instituts wurden 2D-Kristalle einer in ihrer Reaktionsgeschwindigkeit verlangsamten ChR2 Mutante gewonnen, mit denen die Projektionsstruktur des Proteins bestimmt werden konnte [1]. Durch Bestrahlen der 2D-Kristalle im Elektronenmikroskop (EM) mit Laserlicht und noch vor dem Einfrieren konnte der offene Zustand im ChR2-Photozyklus eingefangen werden. Projektions-Differenzkarten zeigten lichtinduzierte Konformationsänderungen beim Übergang vom geschlossenen in den ionenleitenden offenen Zustand, insbesondere eine Neuorientierung der Helices 2, 6 und 7.
Der Substrat/Produkt Antiporter CaiT gehört zur BCCT Familie sekundärer Transporter. Im Gegensatz zu den meisten anderen Transportern ist CaiT nicht ionenabhängig. Die Struktur von CaiT wurde von uns bereits 2010 bestimmt [2]. Mutationen auf der Grundlage der Kristallstrukturen von CaiT aus den Mikroben Escherichia coli und Proteus mirabilis zeigten, dass ein Ersatz der Aminosäure Arginin an Position 262 (R262) durch Alanin den Transport abhängig von Natriumionen (Na+) macht. Strukturelle und biochemische Untersuchungen ergaben, dass R262 eine entscheidende Rolle bei der Substratbindung spielt. Verglichen mit den offenen und geschlossenen Zuständen des CaiT-ähnlichen, Na+-abhängigen Symporters BetP, der in Christine Zieglers Gruppe untersucht wird [3], zeigte sich, wie die abwechselnde Orientierung einer Arginin-Seitenkette den Effekt des Natriumions imitiert [4].
Natrium-Protonen Antiporter gehören schon seit vielen Jahren zum Forschungsprogramm der Abteilung. Elektronenkristallographische Studien des Na+/H+ Antiporters MjNhaP1 aus dem thermophilen Archaeon Methanocaldoccocus jannaschii wurden fortgesetzt. Dabei wurden Helixbewegungen im Umfang von einigen Ångström an der Ionenbindungsstelle als Reaktion auf steigende Na+-Konzentrationen beobachtet. Dagegen änderten pH-Unterschiede in Abwesenheit von Salz die Struktur kaum. Auf der Grundlage des 3D-Modelles von MjNhaP1 konnten die beobachteten Bewegungen einzelnen Helices zugeordnet werden.
2. Membranproteinkomplexe in Mitochondrien, Chloroplasten und Prokaryonten
Struktur des Respirasoms. Durch Einzelpartikel-Kryo-Elektronenmikroskopie wurde eine 3D-Karte mit einer Auflösung von rund 20 Å der Atmungskette des Superkomplexes I1III2IV1 aus einem Mitochondrium des Rindenherzens gewonnen, der auch als Respirasom bezeichnet wird. Die Karte zeigt, wie die drei Protonenpumpen der inneren Mitochondrienmembran zusammenwirken, um in der Atmungskette Elektronen von NADH auf molekularen Sauerstoff zu übertragen. Mithilfe von Kryo-ET konnte gezeigt werden, dass in Säugermitochondrien ein großer Teil des Komplexes I in Superkomplexen der inneren Membran organisiert ist.


Struktur der mitochondrialen ATP-Synthase in situ. Die Organisation der mitochondrialen ATP-Synthase in der inneren Membran gehört zu den zentralen Forschungsinteressen der Gruppe. Nach der Entdeckung, dass die ATP-Synthase in Säugermitochondrien in bis zu 1 µm langen Dimer-Reihen entlang der stark gebogenen Cristae-Ränder angeordnet ist, stellte sich heraus, dass diese Anordnung in allen anderen untersuchten Organismen, einschließlich Pilzen und Pflanzen, im Wesentlichen gleich ist [5]. Eine Mittelung der in situ Dimere aus Hefemitochondrien ergab eine 3D Karte bei 37 Å Auflösung, in die die vorhandenen Röntgenstrukturen eingepasst werden konnten (Abb. 1; [6]).
Die Protonenpumpen der mitochondrialen Atmungskette befinden sich in den Membranbereichen an beiden Seiten der Dimer-Reihen [5], wo sie Protonen in den weitgehend abgeschlossenen Cristae-Raum abgeben. Diese konservierte Anordnung von Dimer-Reihen und Protonenpumpen in der Cristae ist ein grundlegendes Merkmal der Membranarchitektur in normalen, aktiven Mitochondrien. Es wird angenommen, dass die Cristae als Protonenfallen fungieren, in denen Protonen von ihrer Quelle an den Atmungskettensuperkomplexen zu den Protonenabflüssen an den Dimer-Reihen fließen (Abb. 2). Diese Anordnung könnte für die effiziente Synthese von ATP in den Mitochondrien notwendig, mindestens aber günstig sein.

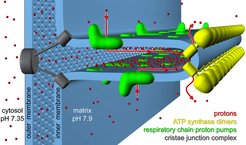
Abb. 2: Schematische Darstellung der Anordnung von drei großen Membranproteinkomplexen in der Cristae der inneren Mitochondrienmembran.
Änderung der mitochondrialen Membranstruktur in alternden Zellen. Die hoch konservierte Anordnung der ATP-Synthase in der inneren Mitochondrienmembran ändert sich grundlegend in seneszenten Kulturen des Fadenpilzes Podospora anserina, wie durch Kryo-ET entdeckt wurde (Abb. 3; [7]). Mit zunehmendem Alter werden die Cristae flacher, die ATP-Synthase Dimere zerfallen und am Ende bildet die Mitochondrien-Matrix Vesikel. In den Matrixvesikeln liegt die ATP-Synthase nur noch monomer vor. Die lokale Membrankrümmung, verglichen mit der von intakten Cristae, ist minimal. Sollte die Hypothese zutreffen, dass die Cristae für eine effektive ATP-Produktion erforderlich sind, dann würden diese Mitochondrien nicht mehr in der Lage sein, genügend ATP in den alternden Zellen bereit zu stellen.


Elektronen Kryo-ET von Thylakoidmembranen aus Chloroplasten zeigte, dass im Gegensatz zu Mitochondrien die Chloroplasten-ATP-Synthase keinerlei Dimere oder Reihen bildet, sondern auf die flachen Membranregionen der Grana-Endmembranen oder Stromalamellen beschränkt ist. Die unterschiedliche Anordnung der ATP-Synthase in den beiden Membransystemen von Mitochondrien und Chloroplasten könnte die unterschiedlichen pH-Gradienten in Chloroplasten-Thylakoiden und Mitochondrien widerspiegeln: Die große Differenz des pH-Werts zwischen dem Thylakoid-Lumen und dem Chloroplasten-Stroma macht vielleicht eine aufwendige Anordnung der ATP-Synthase, wie sie in den mitochondrialen Cristae vorliegt, unnötig.
Projektgruppe Janet Vonck
Die Gruppe untersucht Proteinkomplexe mittels Elektronenmikroskopie und Einzelpartikel-Elektronen-Kryo-Mikroskopie.
ATP-Synthasen. Die A-Typ ATP-Synthase der Archaeen unterscheidet sich von den F1Fo Komplexen in Bakterien, Mitochondrien und Chloroplasten. In Zusammenarbeit mit Volker Müller, Universität Frankfurt, wurde die A-Typ ATP-Synthase aus der hyperthermophilen Archaeenart Pyrococcus furiosus mittels Einzelpartikel-Elektronenmikroskopie untersucht. Eine Rekonstruktion von negativ gefärbten Bildern bei 23 Å Auflösung zeigte deutlich die beiden peripheren Stiele. Röntgenstrukturen von A-Typ ATPase-Untereinheiten wurden in die EM-Karte eingepasst, wodurch sich ein nahezu vollständiges Modell des Komplexes ergab. Dank Kryo-EM wurde die Karte bis zu einer Auflösung von derzeit 12,5 Å verbessert.
F420-abhängige Hydrogenase Frh. In Zusammenarbeit mit der Gruppe von Seigo Shima, Max-Planck-Institut für terrestrische Mikrobiologie, Marburg, wird die Struktur der F420-abhängigen NiFe Hydrogenase untersucht. Frh, ein Schlüsselenzym der Methanogenese, besteht aus drei verschiedenen Untereinheiten, die in einem 1,2 MDa Komplex mit tetraedrischer Symmetrie angeordnet sind. Zunächst wurde ein Datensatz von 84.000 Partikeln auf Film gesammelt. Daraus wurde eine Kryo-EM Karte des Komplexes bei einer Auflösung von etwa 4,5 Å errechnet. Zwei Untereinheiten sind homolog zu den großen und kleinen Untereinheiten der bekannten bakteriellen NiFe-Hydrogenase. Die dritte Untereinheit, die einen FAD-Kofaktor bindet und die Substratbindungsstelle enthält, wurde de novo modelliert und zeigte eine neuartige Faltung. Mit dem neuen Falcon CMOS Elektronendetektor wurde ein neuer Datensatz von 30.000 Teilchen gesammelt. Die Kombination der verbesserten Abbildungseigenschaften des Detektors und eine Korrektur von strahlungsinduzierten Objektbewegungen ermöglichte eine deutliche Verbesserung im Vergleich zu Film. Die neue Struktur hat nunmehr eine Auflösung von 3,36 Å mit guter Dichte für die Darstellung und Position der meisten Seitenketten (Abb. 4; [8]).
![Abb. 4: Die NiFe Hydrogenase Frh. Links: Kryo-EM Karte mit jedem Heterotrimer in einer anderen Farbe. Rechts: Modell eines Hetereotrimers. Die drei Untereinheiten sind in verschiedenen Farben gezeichnet. Jedes Heterotrimer enthält eine Elektronenübertragungskette, bestehend aus einem NiFe Zentrum (grüne und orange Kugeln), vier [4Fe4S]-Clustern (orange und gelb) und einem FAD-Cofaktor.](/11606473/original-1508157246.jpg?t=eyJ3aWR0aCI6MzQxLCJmaWxlX2V4dGVuc2lvbiI6ImpwZyIsIm9ial9pZCI6MTE2MDY0NzN9--8b0c627a0ac231bb644da1f10df0b903c5c10dba "Abb. 4: Die NiFe Hydrogenase Frh. Links: Kryo-EM Karte mit jedem Heterotrimer in einer anderen Farbe. Rechts: Modell eines Hetereotrimers. Die drei Untereinheiten sind in verschiedenen Farben gezeichnet. Jedes Heterotrimer enthält eine Elektronenübertragungskette, bestehend aus einem NiFe Zentrum (grüne und orange Kugeln), vier [4Fe4S]-Clustern (orange und gelb) und einem FAD-Cofaktor.")
![Abb. 4: Die NiFe Hydrogenase Frh. Links: Kryo-EM Karte mit jedem Heterotrimer in einer anderen Farbe. Rechts: Modell eines Hetereotrimers. Die drei Untereinheiten sind in verschiedenen Farben gezeichnet. Jedes Heterotrimer enthält eine Elektronenübertragungskette, bestehend aus einem NiFe Zentrum (grüne und orange Kugeln), vier [4Fe4S]-Clustern (orange und gelb) und einem FAD-Cofaktor.](/11606473/original-1508157246.jpg?t=eyJ3aWR0aCI6MjQ2LCJvYmpfaWQiOjExNjA2NDczfQ%3D%3D--8c97dfd6caa333b3c164ef7d6f69b03216381d29)
Projektgruppe Özkan Yildiz
Die Gruppe beschäftigt sich hauptsächlich mit der Struktur und Funktion von Proteinen und Proteinkomplexen des Membrantransports und der Signalübertragung. Die wichtigste Methode ist die Röntgen-Kristallographie von 3D-Kristallen.
Bakterielle Toxine. Das durch Lebensmittel übertragbare Bakterium Listeria monocytogenes ist ein schnell wachsender, fakultativ intrazellulärer Krankheitserreger. Lysteriolysin O (LLO), ein Mitglied der Cholesterin-abhängigen Cytolysin (CDC)-Familie, ist ein porenbildendes Toxin. In Zusammenarbeit mit der Gruppe von Trinad Charkaborty, Universität Gießen, wurde vor kurzem die 2,4 Å Kristallstruktur von LLO in seiner löslichen, monomeren Form aufgeklärt. Die Gesamtstruktur von LLO ähnelt der von Perfringolysin O (PFO). Das lösliche Toxin fügt sich zu 35-50 nm großen Ringen aneinander und bildet auf diese Art große Poren in der Zellmembran. Die Gruppe ermittelte auch die 2,5 Å Kristallstruktur des verwandten Pneumolysin (PLY) von Streptococcus pneumoniae, einem wichtigen Krankheitserreger beim Menschen. Wie LLO bildet auch PLY Reihen von Molekülen, die im Kristall Seite an Seite ausgerichtet sind, was einer Anordnung vor der Porenbildung ähneln könnte.
Forschungsgruppe Thomas Meier
Der Forschungsschwerpunkt der Gruppe liegt auf dem Gebiet der ATP-Synthasen, die in allen Organismen der Hauptlieferant von Adenosintriphosphat (ATP) sind. Der Komplex ist eine Art molekularer Motor, bestehend aus einem Stator und einem Rotor. In ihrer 6-jährigen Forschungstätigkeit am Institut konzentrierten sich die Wissenschaftler auf die strukturelle und funktionelle Charakterisierung der Rotorringe im Fo -Komplex, den Fo -Komplex selbst sowie das Design der ATP-Synthase als molekularer Motor. Ein Highlight war die Ermittlung der Strukturen von mehreren Protonen- oder Na+-bindenden Rotorringen bei hoher Auflösung. Die Ergebnisse dieser Studien lieferten einzigartige Einblicke in die Funktion der ATP-Synthase als molekulare Maschine [9].
Forschungsgruppe Daniel Rhinow
Die Gruppe konzentriert sich auf die Herstellung, Analyse und biotechnologische Anwendungen künstlicher Membranen aus synthetischen und biologischen Bausteinen. Darüber hinaus verwenden die Forscher biologische Membranen als Modellsysteme, um deren zeitliche und räumliche Organisation zu verstehen. Die Elektronenmikroskopie wird zur Strukturanalyse von biologischen und künstlichen Membranen verwendet. Ebenso nutzt die Gruppe ihre Erfahrungen aus der Nanotechnologie, um neue Methoden für die Elektronenmikroskopie zu entwickeln, wie zum Beispiel elektrostatische Phasenplatten [10] und neue Materialien für Trägerfilme.
Forschungsgruppe Christine Ziegler
Die Forschung in der Gruppe von Christine Ziegler konzentriert sich auf das molekulare Verständnis des Membrantransports von osmolytisch aktiven Stoffen, Aminosäuren und Ionen in Bakterien und Säugern, beispielsweise in der Niere. In der Vergangenheit untersuchten die Wissenschaftler vor allem ionengekoppelte Sekundärtransporter. Die Arbeit an dem Betain-Transporter BetP aus Corynebacterium glutamicum ergab grundlegende Einblicke in den Prozess des ionengekoppelten Osmolyttransports. Durch die Aufklärung mehrerer atomarer Strukturen von BetP wurden acht funktionell relevante Konformationen sowie die inaktive und aktive Konformation von BetP beobachtet [3]. Die Fülle an strukturellen, funktionellen und Bioinformatik-Daten für einen einzigen Transporter ermöglichte es, den detaillierten Mechanismus der Substrat/Co-Substrat-Kopplung zu beschreiben.
Literaturhinweise