Forschungsbericht 2013 - Max-Planck-Institut für Dynamik komplexer technischer Systeme
Neue rechnergestützte Methoden zur Aufklärung von zellulären Signaltransduktionswegen
Zelluläre Netzwerke
Zellen bestehen aus einer unvorstellbar großen Menge an unterschiedlichen und komplexen biomolekularen Komponenten. Dazu gehören u.a. bis zu 100.000 verschiedene Proteine, Membranen, DNA, RNA und Tausende von Stoffwechselprodukten (Metaboliten). Während diese Bestandteile das molekulare Erscheinungsbild der Zelle prägen, sind es gerade ihre gegenseitigen Interaktionen, welche Dynamik und – wörtlich – Leben in die Zellen bringen. Interaktionen von zellulären Komponenten lassen sich abstrakt gut in Form von Netzwerken beschreiben: Die Knoten repräsentieren die beteiligten Moleküle und die Kanten zwischen ihnen die stattfindenden Interaktionen. Je nach Typ der Knoten und Interaktionen kann man verschiedene Klassen von biologischen Netzwerken unterscheiden. In Stoffwechselnetzwerken repräsentieren die Knoten Metabolite (z. B. Zucker) und die Kanten die zwischen ihnen ablaufenden biochemischen Reaktionen (z. B. Reaktionen der Glykolyse). In genregulatorischen und Signaltransduktionsnetzwerken repräsentieren die Knoten dagegen meistens Proteine oder Gene und die Kanten Aktivierungs- oder Inhibierungsprozesse, über die sich die beteiligten Partner gegenseitig in ihrer Aktivität beeinflussen.
Interaktionen in Signaltransduktionsnetzwerken oft nur teilweise bekannt
Während man die Struktur von metabolischen Netzwerken bereits für viele Organismen als weitgehend aufgeklärt betrachtet, sind Signaltransduktionsnetzwerke (STN) weit weniger gut charakterisiert. Diese Netzwerke haben im Wesentlichen die Aufgabe, externe und interne Signale der Zelle zu erfassen, zu prozessieren und entsprechende Reaktionen oder Antworten einzuleiten. Die Signalverarbeitung erfolgt in der Regel durch kommunizierende Proteine, die sich z. B. gegenseitig phosphorylieren können, und endet oft mit dem An- oder Ausschalten der Transkription (Expression) bestimmter Gene. Ein klassisches Beispiel für ein STN ist das Binden des Hormons Insulin an den extern zugänglichen Insulinrezeptor einer Zelle, wodurch interne Signalkaskaden ausgelöst werden, die letztlich den Zuckerstoffwechsel der Zelle regulieren. STN werden typischerweise in Netzwerk-Karten dargestellt, die auch in verschiedenen Online-Datenbanken zugänglich sind. Diese „kanonischen” Karten können aber nur als eine Art grobe Vorlage verstanden werden, denn ein Signalweg hat gewöhnlich variierende Ausprägungen in verschiedenen Zelltypen, und es sind oft nicht alle seiner Interaktionen bekannt. In krankhaften Zellen (wie bei Krebs) sind es zudem gerade Abweichungen der normalen Struktur und Funktion von STN, die zum Verhängnis für einen ganzen Organismus werden können. Die Entwicklung mathematischer Methoden zur Rekonstruktion der Struktur von STN aus experimentellen Daten ist daher ein wichtiges Forschungsgebiet der Systembiologie.
Das Prinzip der Netzwerkrekonstruktion


Angetrieben wird die Entwicklung theoretischer Ansätze zur biologischen Netzwerkrekonstruktion (network reconstruction, network inference, oder reverse engineering) durch die rapide wachsende Maschinerie experimenteller Hochdurchsatz-Messverfahren in der Molekularbiologie. Diese Technologien können die großen Mengen an Daten generieren, die für eine datengetriebene Rekonstruktion zellulärer Netzwerke benötigt werden. So lassen sich z. B. die Aktivitäten zahlreicher Netzwerkkomponenten (wie Protein-Phosphorylierungen oder Genexpressionen) nach einer Anregung eines STN zeitaufgelöst verfolgen. Rekonstruktionsalgorithmen versuchen nun aus diesen Daten die zugrundeliegenden Interaktionen zwischen den Knoten abzuleiten (Abb. 1). Dabei kann man zwei Klassen von Ansätzen unterscheiden: solche, die bekanntes Vorwissen über die Netztopologie mit eingehen lassen und solche, die ein Netzwerk ohne Vorgaben rekonstruieren. Die ARB-Gruppe („Analyse und Redesign Biologischer Netzwerke”) am Max-Planck-Institut für Dynamik komplexer technischer Systeme in Magdeburg beschäftigt sich seit Längerem mit der Entwicklung und Anwendung verschiedener theoretischer Methoden der Netzwerkrekonstruktion [1−4]. Im Folgenden soll beispielhaft ein jüngst publiziertes Verfahren genauer vorgestellt werden.
Der SigNetTrainer-Ansatz
Die SigNetTrainer-Methode [1] wurde in Zusammenarbeit mit Partnern von der Technischen Universität in Athen entwickelt und verwendet als Ausgangspunkt einen Interaktionsgraphen (IG), der die bekannten Knoten und Interaktionen im zu untersuchenden STN abbildet. Aktivierende Interaktionen werden dabei über positive und inhibierende über negative Kanten erfasst (Abb. 2A). IG sind kanonische Darstellungsformen von STN in Lehrbüchern, Publikationen und Datenbanken und eignen sich somit auch ideal zur Repräsentation des Vorwissens im Rechner. Wie für alle Netzwerkrekonstruktionsmethoden werden darüber hinaus Messdaten aus möglichst vielen Experimenten benötigt. Idealerweise werden dabei verschiedene Knoten (auch in Kombination) angeregt, z. B. indem relevante externe Signale (wie Hormone oder Wachstumsfaktoren) auf das Netzwerk gegeben werden oder andere Knoten im Netz durch spezifische Inhibitoren oder auch durch Gen-Knockouts in ihrer Aktivität unterdrückt werden. Daraufhin wird der resultierende Aktivierungszustand der (messbaren) Proteine im Netz gemessen. Im vorliegenden Beispiel (Abb. 2) handelt es sich um ein Netz mit sieben Knoten, davon können zwei (A und D) angeregt werden, während der Zustand von drei anderen (F, G, H) gemessen werden kann. In drei verschiedenen Experimenten (E1−E3) wurden A und D in unterschiedlichen Kombinationen angeregt. Die Methode erfordert, dass die Daten diskretisiert werden, d. h. man erfasst für jeden gemessenen Knoten ob seine Aktivität im Vergleich zum Ausgangszustand hoch- (+1) oder runterreguliert (−1) wurde, oder ob keine Veränderung (0) aufgetreten ist.

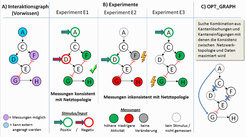
Obwohl ein Interaktionsgraph ein denkbar einfaches Modell eines STN ist, schränkt es seine möglichen qualitativen Verhaltensweisen bereits nachhaltig ein, und so kann es passieren, dass einige experimentelle Befunde inkonsistent mit der vorgegebenen Netzwerkstruktur sind. Als eine notwendige Konsistenzregel kann man erwarten, dass wenn sich die Vorzeichen der diskretisierten Daten nach der Anregung eingepegelt haben, eine festgestellte Veränderung eines Knotenzustandes (+1 oder −1) kausal durch eine passende Änderung eines Vorgängerknotens erklärt werden muss (außer wenn der Knoten selbst direkt angeregt wurde). Das Experiment E1 in Abbildung 2 folgt dieser Regel: Die Aktivitätssteigerung in F kann durch das positive Eingangssignal in A, welches über (nicht gemessenes) C an F weitergegeben wird, erklärt werden. Da E den Knoten G negativ beeinflusst (negative Kante), erklärt die reduzierte Aktivität von E (verursacht durch das negative Inputsignal in D) die Aktivitätssteigerung in G und H. Im Falle der Experimente E2 und E3 wird die Konsistenzregel dagegen verletzt. In E2 sollte F seine Aktivität verringern, wenn D (und dadurch E) extern aktiviert wird. In E3 erwarten wir eine reduzierte statt erhöhte Aktivierung in G und H, da der einzige Pfad von A nach G (A→C→D→E–|G) negativ ist und somit ein negatives Signal vom positiv angeregten A zu G und H fließen sollte.
Als ein wichtiges Ergebnis konnte die ARB-Gruppe zeigen, dass sich die aus der Konsistenzregel ergebenden Abhängigkeiten zwischen den Knoten eines IG als ein System von linearen Ungleichungen über Variablen mit ganzzahligem Wertebereich formulieren lassen [1]. Mit diesen Ungleichungen als Nebenbedingungen lassen sich dann Optimierungsprobleme formulieren, mit denen Inkonsistenzen zwischen Netzwerkstruktur und experimentellen Daten detektiert und schließlich – durch geeignete Veränderungen in der Netzwerktopologie – minimiert werden können. Eine Operation zur Optimierung der Netzwerkstruktur ist OPT_SUBGRAPH, wo optimale Kombinationen von zu entfernenden Kanten ermittelt werden, die den Fehler zwischen Daten und Netzwerk über alle Experimente minimieren. Noch einen Schritt weiter geht OPT_GRAPH, wo Kombinationen aus Entfernungen existierender und Einfügungen neuer Kanten gesucht werden, die zu maximaler Konsistenz zwischen Daten und Netzwerktopologie führen. Abbildung 2C zeigt eine solche optimale Lösung: durch Löschen der Kanten C→D und E–|F bei gleichzeitigem Hinzufügen der Kante A→G erreicht man eine Netzwerkstruktur, die nun mit allen experimentellen Befunden konsistent ist. Da in unterbestimmten Systemen mehrere optimale Lösungen existieren können, wurden die Algorithmen so konzipiert, diese vollständig zu bestimmen (sofern die Rechenzeit in sehr großen Netzen nicht zu hoch wird). Alle Algorithmen wurden in einer MATLAB-Toolbox SigNetTrainer implementiert, in der IBM CPLEX OPTIMIZER als Löser für die aufgestellten ganzzahligen Optimierungsprobleme verwendet wird. Die aufgestellten Probleme können leicht mehrere Tausend Variablen und Ungleichungen enthalten.
Anwendung: die sbv IMPROVER challenge
In einer ersten Studie wurde SigNetTrainer eingesetzt, um zelltyp-spezifische Eigenschaften des STN des epidermalen Wachstumsfaktors in Hepatozyten (häufigster Zelltyp in der Leber) zu bestimmen [1]. Es konnten wichtige Schlussfolgerungen zu offensichtlich inaktiven bzw. fehlenden Interaktionen gezogen werden.
Wie SigNetTrainer wurden in den letzten 10 bis 15 Jahren zahlreiche neue Methoden zur biologischen Netzwerkrekonstruktion vorgeschlagen. Um diese Methoden unabhängig zu evaluieren, wurden Initiativen wie DREAM [5] und sbv IMPROVER [6] angestoßen. Diese Plattformen präsentieren auf ihren Webseiten in regelmäßigen Abständen typische biologische Rekonstruktionsprobleme mit realen oder simulierten Datensätzen, die dann durch die teilnehmenden Teams mit ihren Algorithmen gelöst werden können. Die rekonstruierten Netzwerke müssen bis zu einem vorgegebenen Termin eingereicht werden, ehe sie vom Organisationskomitee mittels bekannter Goldstandards und anderer Evaluationskriterien bewertet werden. Die Teams mit den besten Resultaten werden bekannt gegeben und ausgezeichnet.
![Abb. 3: Übersicht über die 4. Sub-Challenge („Species Specific Network Inference”) der sbv IMPROVER „Species Translation challenge” (siehe auch [7]). Zentrale Aufgabe für die Teilnehmer der Challenge war es, spezifische STN des Menschen bzw. der Ratte anhand von umfassenden Protein-Phosphorylierungs- und Genexpressionsdaten zu bestimmen. Als Ausgangspunkt war ein Referenznetzwerk mit bekannten Interaktionen vorgegeben.](/11601625/original-1508156977.jpg?t=eyJ3aWR0aCI6ODQ4LCJmaWxlX2V4dGVuc2lvbiI6ImpwZyIsIm9ial9pZCI6MTE2MDE2MjV9--a6a4b901abdf44e29b60968e3401287d2e2204cd "Abb. 3: Übersicht über die 4. Sub-Challenge („Species Specific Network Inference”) der sbv IMPROVER „Species Translation challenge” (siehe auch [7]). Zentrale Aufgabe für die Teilnehmer der Challenge war es, spezifische STN des Menschen bzw. der Ratte anhand von umfassenden Protein-Phosphorylierungs- und Genexpressionsdaten zu bestimmen. Als Ausgangspunkt war ein Referenznetzwerk mit bekannten Interaktionen vorgegeben.")
![Abb. 3: Übersicht über die 4. Sub-Challenge („Species Specific Network Inference”) der sbv IMPROVER „Species Translation challenge” (siehe auch [7]). Zentrale Aufgabe für die Teilnehmer der Challenge war es, spezifische STN des Menschen bzw. der Ratte anhand von umfassenden Protein-Phosphorylierungs- und Genexpressionsdaten zu bestimmen. Als Ausgangspunkt war ein Referenznetzwerk mit bekannten Interaktionen vorgegeben.](/11601625/original-1508156977.jpg?t=eyJ3aWR0aCI6MjQ2LCJvYmpfaWQiOjExNjAxNjI1fQ%3D%3D--8ed519e15044074f7d834cc576041a8cc5a90aba)
Unter Verwendung der SigNetTrainer-Methode nahm ein Team aus Forschern der ARB- und Prozesstechnik-Gruppe des MPI Magdeburg an der 4. Sub-Challenge der jüngsten sbv IMPROVER „Species Translation Challenge” teil [7]. Zentrale Aufgabe für die Teilnehmer dieser Sub-Challenge war es, spezifische STN von bronchialen Epithelzellen im Menschen bzw. in der Ratte anhand von umfassenden Protein-Phosphorylierungs- und Genexpressionsdaten aus 26 Stimulus-Response-Experimenten zu bestimmen (Abb. 3). Da als Ausgangspunkt ein Referenznetzwerk mit bekannten (bzw. hypothetischen) Interaktionen vorgegeben war, handelte es sich um einen idealen Anwendungsfall für den SigNetTrainer-Ansatz. Die große Herausforderung für alle Teilnehmer war allerdings die enorme Größe des betrachteten Netzwerks (mehr als 200 Knoten und 500 Kanten) und der riesige Datensatz mit mehr als 2.000 diskretisierten Datenpunkten, jeweils für Mensch und Ratte. Mit einigen algorithmischen Anpassungen und Vereinfachungen war es dem MPI-Team aber möglich, die Methodik auf dieses großskalige System anzuwenden und somit jeweils für beide Spezies eine Netzwerktopologie zu ermitteln, welche Inkonsistenzen zwischen Daten und Referenznetzwerk minimiert. Dabei wiesen die Ergebnisse darauf hin, dass viele Interaktionen in den Referenzsignalwegen offensichtlich nicht aktiv waren, insbesondere an solchen Stellen, wo Proteine der Signaltransduktion mit Genen verknüpft waren. Letztlich wurde das MPI-Team „Reconstructors” mit seiner SigNetTrainer-Methode zusammen mit einem weiteren Team als beste Performer dieser Sub-Challenge ausgezeichnet.
Ausblick
Der oben beschriebene Erfolg unterstreicht das Potenzial der SigNetTrainer-Methode und motiviert deren Einsatz in weiteren konkreten Anwendungen. Dazu etablierte die ARB-Gruppe mehrere Kooperationen bzw. Projekte mit experimentellen Partnern. Zwei Beispiele sind die Aufklärung der veränderten Topologien im menschlichen JAK (Januskinase)-Signalweg in bestimmten Krankheitsfällen (mit Gruppen von der Otto-von-Guericke-Universität Magdeburg) und die Rekonstruktion von genregulatorischen Netzen im Stäbchenbakterium Pseudomonas aeruginosa, einem gefürchteten humanen Krankheitserreger (mit Susanne Häußler, Helmholtz-Zentrum für Infektionsforschung in Braunschweig). Dabei werden auch andere in der ARB-Gruppe entwickelte Rekonstruktionsalgorithmen zum Einsatz kommen, u.a. solche, die nicht auf ein Referenznetzwerk zurückgreifen müssen [2,4] oder genetische Variationen als natürliche Anregungen zur Identifikation von Netzwerken nutzen [3]. Außerdem sollen verstärkt auch hybride Ansätze zum Einsatz kommen, bei denen qualitative/statische (wie beim SigNetTrainer) mit quantitativen/dynamischen Modellbeschreibungen (Differentialgleichungen) vereint werden, um möglichst den gesamten Informationsgehalt experimenteller Daten für die Netzwerkrekonstruktion ausnutzen zu können. Ein wichtiges Augenmerk bleibt dabei immer die Skalierbarkeit der Algorithmen, um letztlich mit der sich ergebenden hohen kombinatorischen Komplexität in großen biologischen Netzwerken umgehen zu können.