Forschungsbericht 2012 - Max-Planck-Institut für Dynamik komplexer technischer Systeme
Molekulare Komplexität in Chemie und Biologie
Wasserstofferzeugung in der Natur und im Reagenzglas
Molekularer Wasserstoff (H2) ist ein Energieträger der Zukunft, aber schwierig zu erzeugen und zu handhaben. Er weist die stabilste aller homonuklearen chemischen Einfachbindungen auf, was ihn so aufwendig in der Erzeugung, aber so attraktiv als Speicher macht. In der Natur erzeugen Enzyme aus Mikroorganismen – die Hydrogenasen für Energie– Wasserstoff bei Raumtemperatur und bei normalem atmosphärischem Druck. Wenn es gelingt, ihren Reaktionsmechanismus im Detail zu entschlüsseln, die wichtigsten Schlüsselkomponenten zu identifizieren, die Bauprinzipien der Natur zu abstrahieren und im Labor nachzubauen, ist die Wissenschaft in der Entwicklung von Prozessen mit alternativen Energieträgern einen großen Schritt weiter. Beispielsweise für den Betrieb von Brennstoffzellen ist hier entweder die Nutzung enzymatisch erzeugten Wasserstoffes möglich oder sogar der Einsatz der Mikroorganismen als biologische Kathode, an der in der Brennstoffzelle Wasserstoff erzeugt wird (mikrobakterielle Brennstoffzelle).
Eine Proteinstruktur verrät nicht alles
, eine Vergößerung von Details des aktiven Zentrum (Mitte) mit einem in der Proteinstruktur nicht eindeutig zuordnenbaren Brückenliganden und ein struktureller Nachbau des aktiven Zentrums des Enzyms (rechts).")
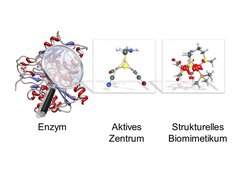
Die Verfügbarkeit einer experimentell bestimmten Struktur von Proteinen beantwortet manchmal nicht alle offenen Fragen. Seit einigen Jahren gibt es z. B. Röntgenstrukturen des Enzyms der Hydrogenasen aus verschiedenen Mikroorganismen, die Wasserstoff erzeugen können. Das aktive Zentrum, an dem die Reaktion stattfindet, ist eine komplizierte Verbindung aus Eisenatomen, Schwefelatomen und kleinen Liganden aus der anorganischen Chemie wie Kohlenmonoxid und Cyanid. Die beiden Eisenatome des aktiven Zentrums sind durch einen verbrückenden Liganden verknüpft, welcher als Zentralatom entweder ein Sauerstoffatom, eine Aminogruppe oder eine Methylgruppe enthalten könnte. Alle besitzen fast die gleiche Anzahl von Elektronen und lassen sich deshalb in der Röntgenstrukturanalyse schwer eindeutig zuordnen. Spektroskopische Untersuchungen am Enzym selbst waren auch nicht eindeutig in der Zuordnung. Ein Modellkomplex mit strukturellen Ähnlichkeiten zum aktiven Zentrum des Enzyms wurde hergestellt und untersucht (Abb. 1). Durch eine Kombination von aufwendigen Methoden der Spektroskopie und der computergestützten Chemie konnten Parameter für ein Stickstoffatom in der Mitte der Brückenliganden gewonnen und interpretiert werden, sodass auch im Enzym die Existenz einer Aminogruppe als gesichert gilt [1].
Klein macht den Unterschied
![Abb. 2: Sauerstofftolerante Hydrogenase aus der Klasse der [NiFe]-Hydrogenase. Die Toleranz gegenüber Luftsauerstoff wird nicht, wie lange angenommen, durch Veränderungen am aktiven Zentrum hervorgerufen. Es ist vielmehr das Auftreten einer neuen Art von Eisen-Schwefel-Clustern in der kleinen Untereinheit, das für die Stabilität des Enzyms in Anwesenheit von Sauerstoff verantwortlich ist. Der neue 4Fe-3S-6-Cysteincluster weist ein ungewöhnliches Redoxverhalten auf und schützt so das aktive Zentrum, welches sich 10 Å entfernt befindet.](/11591754/original-1508156581.jpg?t=eyJ3aWR0aCI6MzQxLCJmaWxlX2V4dGVuc2lvbiI6ImpwZyIsIm9ial9pZCI6MTE1OTE3NTR9--4576b7aeb54615fffe82901a4d40131dab45e1b4 "Abb. 2: Sauerstofftolerante Hydrogenase aus der Klasse der [NiFe]-Hydrogenase. Die Toleranz gegenüber Luftsauerstoff wird nicht, wie lange angenommen, durch Veränderungen am aktiven Zentrum hervorgerufen. Es ist vielmehr das Auftreten einer neuen Art von Eisen-Schwefel-Clustern in der kleinen Untereinheit, das für die Stabilität des Enzyms in Anwesenheit von Sauerstoff verantwortlich ist. Der neue 4Fe-3S-6-Cysteincluster weist ein ungewöhnliches Redoxverhalten auf und schützt so das aktive Zentrum, welches sich 10 Å entfernt befindet.")
![Abb. 2: Sauerstofftolerante Hydrogenase aus der Klasse der [NiFe]-Hydrogenase. Die Toleranz gegenüber Luftsauerstoff wird nicht, wie lange angenommen, durch Veränderungen am aktiven Zentrum hervorgerufen. Es ist vielmehr das Auftreten einer neuen Art von Eisen-Schwefel-Clustern in der kleinen Untereinheit, das für die Stabilität des Enzyms in Anwesenheit von Sauerstoff verantwortlich ist. Der neue 4Fe-3S-6-Cysteincluster weist ein ungewöhnliches Redoxverhalten auf und schützt so das aktive Zentrum, welches sich 10 Å entfernt befindet.](/11591754/original-1508156581.jpg?t=eyJ3aWR0aCI6MjQ2LCJvYmpfaWQiOjExNTkxNzU0fQ%3D%3D--58c60bd6cf72f530239decc2920cab004b4e5503)
Die meisten der Hydrogenasen aus der Familie der [NiFe]-Hydrogenasen sind sehr empfindlich gegenüber der Gegenwart von Luftsauerstoff. In der Evolution sind sie als anaerobe Bakterien ideal angepasst an ihre Lebensbedingungen vor Milliarden von Jahren und kommen heute noch in vulkanischen Umgebungen vor. Sie werden durch Sauerstoff irreversibel geschädigt und sind danach nicht mehr aktiv. Eine besondere Unterart der Hydrogenasen hat sich gemäß ihren symbiotischen Lebensbedingungen an die Anwesenheit von Luftsauerstoff angepasst. Obwohl dieser Fakt seit mehreren Jahren bekannt ist, war die Ursache dieser idealen Anpassung bisher nicht erklärbar. Kürzlich gelang es, die Ursache für die Sauerstofftoleranz aufzuklären. Durch eine Kombination von Methoden der Bioinformatik und der dreidimensionalen Modellierung der Proteinstruktur war es möglich zu zeigen, dass keine Veränderungen am oder in der Nähe des aktiven Zentrums für die Sauerstofftoleranz verantwortlich sind (Abb. 2). Stattdessen ist es ein neues, bisher unbekanntes Koordinationsmuster von Aminosäuren am nächsten Eisen-Schwefel-Cluster in der Elektronentransportkette (in der kleinen Proteinuntereinheit), die die sauerstofftoleranten von den sauerstoffempfindlichen [NiFe]-Hydrogenasen unterscheiden [2]. Der Abstand zwischen dem aktiven Zentrum und dem nächstgelegenen Eisen-Schwefel-Cluster beträgt 10 Ångstrom. Dieser neue, bisher nicht bekannte Typ von 4Fe-6Cystein-Clustern weist ein ungewöhnliches Redoxverhalten auf und schützt so das aktive Zentrum vor Schädigung durch Luftsauerstoff über eine relativ große Entfernung.
Von der Natur ins Reagenzglas
![Abb. 3: Darstellung der Gemeinsamkeiten der aktiven Zentren der [FeFe]-, [NiFe]-Hydrogenasen und von Modellverbindungen. Verankerungen durch Wasserstoffbrückenbindungen mit dem umgebenen Protein sind in orange angedeutet. Die strukturelle Flexibilität in der Modellverbindung stellt eine Herausforderung dar.](/11591762/original-1508156581.jpg?t=eyJ3aWR0aCI6MzQxLCJmaWxlX2V4dGVuc2lvbiI6ImpwZyIsIm9ial9pZCI6MTE1OTE3NjJ9--d14cbe507ed507e490b6307ccdafe568b67ec287 "Abb. 3: Darstellung der Gemeinsamkeiten der aktiven Zentren der [FeFe]-, [NiFe]-Hydrogenasen und von Modellverbindungen. Verankerungen durch Wasserstoffbrückenbindungen mit dem umgebenen Protein sind in orange angedeutet. Die strukturelle Flexibilität in der Modellverbindung stellt eine Herausforderung dar.")
![Abb. 3: Darstellung der Gemeinsamkeiten der aktiven Zentren der [FeFe]-, [NiFe]-Hydrogenasen und von Modellverbindungen. Verankerungen durch Wasserstoffbrückenbindungen mit dem umgebenen Protein sind in orange angedeutet. Die strukturelle Flexibilität in der Modellverbindung stellt eine Herausforderung dar.](/11591762/original-1508156581.jpg?t=eyJ3aWR0aCI6MjQ2LCJvYmpfaWQiOjExNTkxNzYyfQ%3D%3D--9e2894509c321ade43af7e88fa2d0c9275d1801e)
Die Designprinzipien der Natur zu erkennen bedeutet, diese zu abstrahieren und die essenziellen Strukturen auf ein System zu übertragen, das die Natur nachahmt (Biomimetikum). Es gelang das chemische Design von Modellverbindungen, die im Reagenzglas die elektrochemische Reduktion von Protonen zu molekularem Wasserstoff katalysieren. Von einer großtechnischen Umsetzung ist der Prozess aber noch entfernt. Beispielsweise sind zweikernige Komplexe aus Eisenatomen, die Strukturmerkmale des Enzyms aufgreifen und umsetzen, dazu in der Lage (Abb. 3). Die Eisenatome weisen eine Koordinationssphäre auf, die der im Enzym ähnelt. Bei geringer Überspannung setzen sie Wasserstoff frei. Unerlässlich für das Verständnis und die Aufklärung der Arbeitsweise der Modellsysteme sind die Synthese, die Spektroskopie und die Computerberechnungen. Ebenso lassen sich mononukleare Komplexe herstellen, die ohne ein zweites Eisenatom auskommen, welche Wasserstoff durch elektrochemische Protonenreduktion freisetzen [3]. Bei der Untersuchung und Analyse der biomimetischen Verbindungen zeigt sich, dass manchmal die kleinen Modellverbindungen komplizierter sind als die Natur selber. Im Enzym wird das aktive Zentrum durch eine große Anzahl von Wechselwirkungen mit der Proteinmatrix festgehalten und ist so weniger flexibel als kleine Moleküle in Lösung. Diese sind beweglicher und können durch diese strukturelle Flexibilität mehrere konformationelle Zustände erreichen, die alle bei der Analyse und Interpretation berücksichtigt werden müssen.
Proteine im großen Zusammenhang betrachten
 und dreidimensionale Visualisierung in der Proteinstruktur (3D-Mapping).")
In der Systembiologie verschiebt sich der Fokus von der Untersuchung einzelner Proteine hin zur Untersuchung von Proteinen in Netzwerken. Kinetische Parameter für die Aktivierung, für den Substratumsatz sowie für regulatorische Prozesse sind hierfür notwendig, oftmals aber nicht in der Literatur verfügbar. Grundlage sind aber immer molekulare Wechselwirkungen von u. a. Substrat und Enzym oder Protein-Protein-Interaktionen, die im Computer berechnet und analysiert werden. Hiermit verändert sich auch die Betrachtungsweise von der Untersuchung einzelner Proteine hin zur Charakterisierung einer großen Anzahl von Proteinen in einem weiteren Kontext (Abb. 4). Durch den quantitativen Vergleich der dreidimensionalen Wechselwirkungsfelder [4] zwischen einer großen Anzahl von Proteinen lassen sich Aussagen treffen über den Einfluss einzelner Mutationen auf kinetische Parameter, den Vergleich von Enzymen metabolischer Netzwerke in verschiedenen Organismen [5] oder die Variabilität und Unterschiede von der gleichen Enzym- oder Proteinklasse z. B. des Menschen [6]. Der Ansatz ist nicht begrenzt auf Enzyme, sondern lässt sich ebenso anwenden auf Proteine der Maschinerie der Endozytose oder menschliche Grippeviren.
Die Entwicklung von neuen Algorithmen und Werkzeugen zur Simulation auf verschiedenen Zeitskalen wird in Zukunft die Betrachtung weiterer molekularer Grundlagen für komplizierte Vorgänge in der Chemie und Biologie erlauben.