Forschungsbericht 2009 - Max-Planck-Institut für Herz- und Lungenforschung
Bluthochdruck, Schock, Atherosklerose – Fehlregulationen des Blutgefäßsystems
Unablässig wird Blut durch den menschlichen Körper gepumpt. Der Blutkreislauf sichert das Überleben, indem er alle Bereiche des Körpers mit Sauerstoff und Nährstoffen versorgt. Dies ist nur möglich, indem die Durchlässigkeit der Gefäße und die Kontraktionskraft der muskulären Blutgefäßwand ständig an sich ändernde Bedingungen angepasst werden. Kontrahieren kleine Arterien zu stark, so entsteht Bluthochdruck; ist der Kontraktionszustand nicht ausreichend, so fällt der Blutdruck ab, und es kann zu einem Kreislaufschock kommen. Auch Störungen der Durchlässigkeit der Innenschicht von Blutgefäßen führen zu Störungen der Gewebeversorgung, und eine Ablagerung von Fetten wie Cholesterin führt zu entzündlichen Veränderungen der Gefäßwand, die dann in eine Atherosklerose münden.
Die Wand der Blutgefäße besteht vor allem aus elastischen Fasern und Muskelzellen, deren Kontraktionszustand den Durchmesser des Blutgefäßes reguliert. Die Innenseite des Blutgefäßes wird durch eine dünne Zellschicht, das Endothel, ausgekleidet. Die glatte Gefäßmuskulatur und das Endothel stehen in einem ständigen intensiven Austausch. Zusätzlich wird ihre Funktion durch Hormone und Neurotransmitter, die von den Nerven der Gefäßwand freigesetzt werden, reguliert. Die meisten dieser gefäßaktiven Mediatoren wirken über sogenannte G-Protein-gekoppelte Rezeptoren (Abb. 1).
 sowie die nachgeordneten Gq/G11- (blau) und G12/G13-vermittelten (rot) Signalwege.")
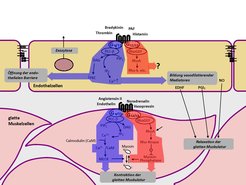
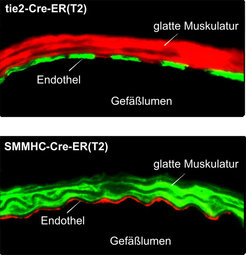
Nach Aktivierung koppeln diese Rezeptoren an G-Proteine, die an der Innenseite der Zellmembran lokalisiert sind und für die Weiterleitung des Signals in das Zellinnere verantwortlich sind. In den Zellen der glatten Gefäßmuskulatur und des Endothels spielen vor allem die G-Proteine Gq/G11 und G12/G13 eine wichtige Rolle, die im Falle von Gq/G11 eine Erhöhung der intrazellulären Ca2+-Konzentration vermitteln und im Falle von G12/G13 den sogenannten Rho/Rho-Kinaseweg aktivieren [1]. Welche Rolle Gq/G11- bzw. G12/G13-vermittelte Signalweiterleitungswege in der Gefäßwand unter normalen und krankhaften Bedingungen spielen, war unbekannt. Da keine Pharmaka für eine spezifische Blockade dieser Signalwege zur Verfügung stehen, entwickelte das Team um Stefan Offermanns Mausmodelle, die eine induzierbare Inaktivierung der Gene der α-Untereinheiten von Gq/G11 und G12/G13 in vivo ermöglichen. Eine derartige konditionale Geninaktivierung wird üblicherweise durch Einsatz des sogenannten Cre/loxP-Systems durchgeführt. Dabei führt die gewebespezifische Expression der Rekombinase Cre zur Inaktivierung des durch loxP-Erkennungssequenzen markierten Gens. Eine derartige gewebespezifische Geninaktivierung kann induzierbar gemacht werden durch die Verwendung einer Cre-Variante, die nur in Anwesenheit des synthetischen Östrogens Tamoxifen aktiv ist. In diesem Falle kommt es erst nach Behandlung der Tiere mit Tamoxifen zur gewebespezifischen Geninaktivierung. Durch Promotoren, die nur in der glatten Muskulatur bzw. nur im Endothel aktiv sind, gelang die induzierbare Geninaktiverung gezielt in einem dieser Gewebe. (Abb. 2).
 sowie in glatten Muskelzellen (SMMHC-CreERT2) exprimieren. Der Nachweis erfolgte durch Verpaarung Cre-transgener Tiere mit einer Cre-Reporterlinie, die ein rot fluoreszierendes Potein (tomato) in nicht rekombinierten Zellen, ein grün fluoreszierendes Protein (EGFP) hingegen in rekombinierten Zellen exprimiert.")
Mittels Mäusen, in denen die Gene von Untereinheiten der G-Proteine Gq/G11 und G12/G13 mit loxP-Erkennungssequenzen markiert waren [2, 3] konnte nun die Rolle dieser zentralen Signalwege in Zellen der Gefäßwand unter normalen und krankhaften Bedingungen untersucht werden.
Molekulare Mechanismen des allergischen Kreislaufschocks
Beim anaphylaktischen Schock handelt es sich um eine besonders schwere und fulminant verlaufende, akut lebensbedrohliche allergische Reaktion, die meistens durch Arzneimittel, Insektengifte oder andere Allergene ausgelöst wird. Die Häufigkeit von anaphylaktischen Reaktionen hat in den letzten Jahrzehnten deutlich zugenommen [4]. Entsprechend sensibilisierte Personen können nach der Zufuhr eines Allergens, das dann die Freisetzung verschiedener Mediatoren aus Mastzellen und basophilen Leukozyten hervorruft, einen anaphylaktischen Schock erleiden. Mediatoren wie Histamin, Proteasen, „Platelet Activating Factor (PAF)“ oder verschiedene Leukotriene und Prostanoide wirken dann lokal sowie systemisch, wobei sie einen dramatischen Abfall des Blutdrucks, eine Störung der endothelialen Barrierefunktion, Herz-Rhythmus-Störungen, ein Absinken der Körpertemperatur, asthmaähnliche Beschwerden sowie diverse Symptome im Verdauungstrakt und an der Haut hervorrufen können. Man nimmt an, dass die Kombination dieser Mediatoreffekte in verschiedenen Organsystemen zum lebensbedrohlichen Krankheitsbild des anaphylaktischen Schocks führt.
Die meisten der bei einem anaphylaktischen Schock gebildeten Mediatoren wirken über Rezeptoren, die an die G-Proteine Gq/G11 und G12/G13 koppeln, auf diverse Körperzellen (wie Immunzellen, Herzmuskelzellen sowie Endothelzellen). Die induzierte Ausschaltung der G-Proteine Gq/G11 und G12/G13 in Endothelzellen hatte keine offensichtlichen Folgen für die normale Funktion des Gefäßsystems. Die Blutdruckregulation sowie der normale Stoffaustausch über die Gefäßwand blieben davon unbeeinflusst. Bei Tieren mit endothelialer Defizienz für den Gq/G11-vermittelten Signalweg führten jedoch verschiedene Mediatoren nicht mehr in der üblichen Weise zu einer Öffnung der endothelialen Barriere. Die systemische Verabreichung des Mediators PAF führt in normalen Tieren zu einem Kreislaufschock-ähnlichen Bild, das meist mit dem Tod der Tiere einhergeht. Interessanterweise waren Gq/G11-defiziente Mäuse gegen diesen Effekt von PAF geschützt. In verschiedenen Modellen des anaphylaktischen Schocks bestätigte sich diese interessante Rolle von Gq/G11 im Endothel der Gefäße (Abb. 3).
![Defekte in der Regulation der endothelialen Barriere in endothelzellspezifischen Gαq/Gα11-defizienten Mäusen. A) Verlauf des Blutdruckes in Wildtypmäusen sowie in Mäusen mit endothelzellspezifischer Gαq/Gα11-Defizienz (EC-q/11-ko; blau) sowie Gα12/Gα13-Defizienz (EC-12/13-ko; rot) nach Gabe des Mediators Histamin. B) Überleben von Wildtypmäusen und endothelzellspezifischen Gαq/Gα11-defizienten Mäusen nach Injektion des anaphylaktischen Mediators PAF. C) Überlebenskurven von Wildtyp-Mäusen und endothelzellspezifischen Gαq/Gα11- und Gα12/Gα13-defizienten Mäuse nach Auslösung einer schweren anaphylaktischen Reaktion. Diese Ergebnisse wurden im Journal of Experimental Medicine publiziert [5].](/338047/original-1293749958.jpg?t=eyJ3aWR0aCI6MzQxLCJmaWxlX2V4dGVuc2lvbiI6ImpwZyIsIm9ial9pZCI6MzM4MDQ3fQ%3D%3D--9b5b9dd79ff950a4c5164c23e4fcc4c3064d2d34 "Defekte in der Regulation der endothelialen Barriere in endothelzellspezifischen Gαq/Gα11-defizienten Mäusen. A) Verlauf des Blutdruckes in Wildtypmäusen sowie in Mäusen mit endothelzellspezifischer Gαq/Gα11-Defizienz (EC-q/11-ko; blau) sowie Gα12/Gα13-Defizienz (EC-12/13-ko; rot) nach Gabe des Mediators Histamin. B) Überleben von Wildtypmäusen und endothelzellspezifischen Gαq/Gα11-defizienten Mäusen nach Injektion des anaphylaktischen Mediators PAF. C) Überlebenskurven von Wildtyp-Mäusen und endothelzellspezifischen Gαq/Gα11- und Gα12/Gα13-defizienten Mäuse nach Auslösung einer schweren anaphylaktischen Reaktion. Diese Ergebnisse wurden im Journal of Experimental Medicine publiziert [5].")
![Defekte in der Regulation der endothelialen Barriere in endothelzellspezifischen Gαq/Gα11-defizienten Mäusen. A) Verlauf des Blutdruckes in Wildtypmäusen sowie in Mäusen mit endothelzellspezifischer Gαq/Gα11-Defizienz (EC-q/11-ko; blau) sowie Gα12/Gα13-Defizienz (EC-12/13-ko; rot) nach Gabe des Mediators Histamin. B) Überleben von Wildtypmäusen und endothelzellspezifischen Gαq/Gα11-defizienten Mäusen nach Injektion des anaphylaktischen Mediators PAF. C) Überlebenskurven von Wildtyp-Mäusen und endothelzellspezifischen Gαq/Gα11- und Gα12/Gα13-defizienten Mäuse nach Auslösung einer schweren anaphylaktischen Reaktion. Diese Ergebnisse wurden im Journal of Experimental Medicine publiziert [5].](/338047/original-1293749958.jpg?t=eyJ3aWR0aCI6MjQ2LCJvYmpfaWQiOjMzODA0N30%3D--796bdfb0887bd107f13cc7720bcc081a4c7836ec)
Die Auslösung einer schweren anaphylaktischen Reaktion, die mit dem Tod von Wildtyp-Tieren einhergeht, hatte in Mäusen mit endothelialer Gq/G11-Defizienz kaum sichtbare Folgen [5]. Diese Tiere waren geschützt gegenüber schwersten anaphylaktischen Reaktionen. Offensichtlich stellt die Aktivierung des Gq/G11-vermittelten Signaltransduktionsweges über verschiedene Rezeptoren auf Endothelzellen den entscheidenden Mechanismus dar, über den es im Rahmen schwerer anaphylaktischer Reaktionen zum Schock mit meist tödlichem Ausgang kommt. Da die Ausschaltung dieses endothelialen Signalweges keinen Einfluss auf die normale Funktion des vaskulären Systems besitzt, jedoch Tiere vor den fatalen Effekten inflammatorischer und anaphylaktischer Mediatoren schützt, stellen diese Signalmoleküle eine interessante neue Zielstruktur für Pharmaka dar, die in der Prophylaxe oder Behandlung anaphylaktischer Reaktionen von Nutzen sein können.
Warum zu viel Salz den Blutdruck erhöht
Mehr als ein Viertel der erwachsenen Weltbevölkerung leidet an Bluthochdruck (Hypertonie), einem der wichtigsten Risikofaktoren für Herz-Kreislauf-Erkrankungen [6]. Einer der ursächlichen Faktoren der Hypertonie ist in vielen Fällen ein erhöhter Salzkonsum, der in den letzten Jahrzehnten immer weiter angestiegen ist und in Industrieländern meist zwischen fünf und zehn Gramm Kochsalz pro Tag liegt (Abb. 4).


Mehr als 80% dieser Salzmenge wird den Nahrungsmitteln während ihrer Herstellung und Verarbeitung zugesetzt, nur eine entsprechend kleine Menge ist in den natürlichen Bestandteilen der Nahrung enthalten oder stammt aus der heimischen Küche.
Der wichtigste Mechanismus, über den der Körper relativ große Salzmengen wieder ausscheidet, besteht in der Erhöhung des Blutdrucks, der die Salz- und Wasserausscheidung durch die Nieren steigert. Während diese der Salz-induzierten Hypertonie zugrunde liegenden Mechanismen gut untersucht sind, ist noch ungeklärt, wodurch der Gefäßwiderstand und damit der Blutdruck nach Salzgabe ansteigt [7]. In dem Verdacht, die salzabhängige Hypertonie hervorzurufen, stehen verschiedene Gefäß-kontrahierende Mediatoren, die über G-Protein gekoppelte Rezeptoren zu einer Kontraktion der Gefäßmuskulatur und damit zur Erhöhung von Gefäßwiderstand und Blutdruck führen.
![A) Verlauf des Blutdruckes in Wildtyp-Tieren sowie nach Induktion einer Gαq/Gα11- (Sm-q/11-ko; blau) bzw. Gα12/Gα13-Defizienz (Sm-13/13-ko; rot) in Mäusen. Der Blutdruck wurde telemetrisch über mehrere Wochen gemessen. B) Verlauf des Blutdruckes in Wildtyp-Tieren sowie in Tieren mit Gαq/Gα11- sowie Gα12/Gα13-Defizienz in der glatten Gefäßmuskulatur vor und nach Auslösung einer salzsensitiven Hypertonie durch Behandlung der Tiere mit dem Mineralokortikoid DOCA sowie einer erhöhten Salzkonzentration im Trinkwasser. Der Blutdruck wurde telemetrisch über mehrere Wochen bestimmt. Diese Ergebnisse wurden in der Zeitschrift Nature Medicine [8] veröffentlicht.](/338159/original-1293750016.jpg?t=eyJ3aWR0aCI6MzQxLCJmaWxlX2V4dGVuc2lvbiI6ImpwZyIsIm9ial9pZCI6MzM4MTU5fQ%3D%3D--34cf7e83af59adf440bbc24dcfbf4c08806f8b42 "A) Verlauf des Blutdruckes in Wildtyp-Tieren sowie nach Induktion einer Gαq/Gα11- (Sm-q/11-ko; blau) bzw. Gα12/Gα13-Defizienz (Sm-13/13-ko; rot) in Mäusen. Der Blutdruck wurde telemetrisch über mehrere Wochen gemessen. B) Verlauf des Blutdruckes in Wildtyp-Tieren sowie in Tieren mit Gαq/Gα11- sowie Gα12/Gα13-Defizienz in der glatten Gefäßmuskulatur vor und nach Auslösung einer salzsensitiven Hypertonie durch Behandlung der Tiere mit dem Mineralokortikoid DOCA sowie einer erhöhten Salzkonzentration im Trinkwasser. Der Blutdruck wurde telemetrisch über mehrere Wochen bestimmt. Diese Ergebnisse wurden in der Zeitschrift Nature Medicine [8] veröffentlicht.")
![A) Verlauf des Blutdruckes in Wildtyp-Tieren sowie nach Induktion einer Gαq/Gα11- (Sm-q/11-ko; blau) bzw. Gα12/Gα13-Defizienz (Sm-13/13-ko; rot) in Mäusen. Der Blutdruck wurde telemetrisch über mehrere Wochen gemessen. B) Verlauf des Blutdruckes in Wildtyp-Tieren sowie in Tieren mit Gαq/Gα11- sowie Gα12/Gα13-Defizienz in der glatten Gefäßmuskulatur vor und nach Auslösung einer salzsensitiven Hypertonie durch Behandlung der Tiere mit dem Mineralokortikoid DOCA sowie einer erhöhten Salzkonzentration im Trinkwasser. Der Blutdruck wurde telemetrisch über mehrere Wochen bestimmt. Diese Ergebnisse wurden in der Zeitschrift Nature Medicine [8] veröffentlicht.](/338159/original-1293750016.jpg?t=eyJ3aWR0aCI6MjQ2LCJvYmpfaWQiOjMzODE1OX0%3D--9236a74c394abb470460143fa0360fcc03cf46f2)
Gefäßverengende Mediatoren, die über derartige Rezeptoren wirken, aktivieren zwei parallele Signalwege in Gefäßmuskelzellen, um eine Kontraktion auszulösen. Einer der beiden Signalwege wird durch die G-Proteine Gq/G11 vermittelt und führt zu einer Erhöhung der freien Ca2+-Konzentration, während der andere Signalweg durch die G-Proteine G12/G13 vermittelt wird und zur Aktivierung des Proteins Rho führt (Abb. 1). Die induzierte Ausschaltung des Gq/G11-vermittelten Signalweges in der glatten Gefäßmuskulatur führte dazu, dass der basale Blutdruck der Tiere deutlich abnahm [8]. Gleichzeitig entwickelten die Tiere jedoch auch keine Hypertonien nach vermehrter Salzgabe (Abb. 5). Wurde hingegen der G12/G13-vermittelte Signalweg ausgeschaltet, blieb der normale Blutdruck unverändert. Allerdings zeigten auch diese Tiere keinen nennenswerten Anstieg ihres Blutdruckes unter salzreicher Ernährung (Abb. 5).
Der Gq/G11-vermittelte Signalweg ist also sowohl für die Aufrechterhaltung des normalen Blutdruckes als auch für die Entwicklung einer salzabhängigen Hypertonie erforderlich, wohingegen interessanterweise der G12/G13-vermittelte Signalweg keine Rolle bei der Aufrechterhaltung des normalen Blutdruckes spielt, jedoch unabdingbar für die Entwicklung einer Salz-induzierten Hypertonie ist [8]. Diese Erkenntnisse stützten die Hypothese, dass in der Tat verschiedene Mediatoren, die G-Protein-gekoppelte Rezeptoren aktivieren, eine zentrale Rolle bei der Auslösung einer salzabhängigen Hypertonie spielen. Da eine Blockade des G12/G13-vermittelten Signalweges keinen Einfluss auf den basalen Blutdruck hat, jedoch den Anstieg des Blutdruckes unter salzreicher Diät verhindert, stellt dieser Signalweg, der aus verschiedenen Komponenten besteht, eine ideale Zielstruktur für neue Blutdruck-senkende Pharmaka dar. Ihre Wirkung sollte auf die Senkung der Hypertonie beschränkt sein und nicht die Gefahr von überschießenden Blutdrucksenkungen, z. B. nach Überdosierung, mit sich bringen – eine Gefahr, die bei vielen derzeit verwendeten Antihypertensiva in Kauf genommen werden muss.