Pathological thickening of the cardiac wall halted
Inhibition of RhoGEF12 leads to considerable improvement in disease progression
The heart responds to the increased stress caused by chronically raised blood pressure, for example, by thickening its wall muscle. In the late stage of this condition, a risk of heart failure arises. Scientists from the Max Planck Institute for Heart and Lung Research have now succeeded in identifying a key molecule in the molecular signalling cascade responsible for this growth. Based on this discovery, they managed to achieve a significant reduction in cardiac wall thickening in animal experiments. In addition, they managed to partly reduce existing thickening of the cardiac wall.
The heart reacts to intensive, long-term stress by increasing its muscle mass. In competitive athletes, this thickening of the cardiac wall is known as athletic heart syndrome or “athlete’s heart”. Whereas in this case, the process is a reversible physiological reaction to physical activity, in other cases, cardiac wall thickening, known medically as cardiac hypertrophy, is a serious condition; its progression frequently leads to death through heart failure. The triggers for this pathological change can include, for example, high blood pressure, arteriosclerosis and cardiac valve defects.
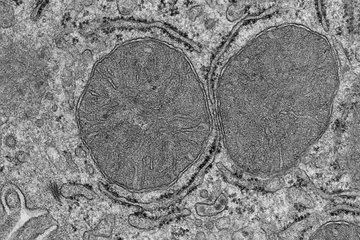
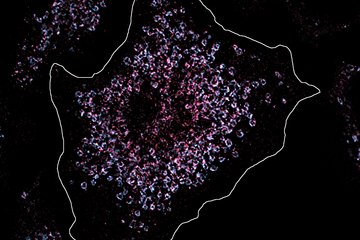
Scientists from the Max Planck Institute for Heart and Lung Research in Bad Nauheim have now identified a crucial interface in the signalling cascade that controls the emergence of cardiac hypertrophy at molecular level. The interface in question is a molecule called RhoGEF12. The crucial indicator was revealed to the researchers in studies on mice, in which the aorta had been artificially narrowed, thereby triggering the development of hypertrophy. “We observed a clear increase in RhoGEF12 activation in the cardiac muscle cells of these mice, “ said Nina Wettschureck, who carried out the study in collaboration with Mikito Takefuji. The Max Planck researchers then used genetically modified mice in which RhoGEF12 could be switched off in cardiac muscle cells in their hypertrophy model. “Four weeks after the beginning of the treatment, the cardiac wall thickening in these mice was clearly less advanced than in animals with RhoGEF12,” explains Wettschureck. In addition, the heart pump output of the mice without RhoGEF12 was considerably better than that in the control group. This led to a higher survival rate in the long term.
An answer to the question as to whether existing hypertrophy can be reversed by switching off RhoGEF12 was important from a clinical perspective. The Bad Nauheim researchers therefore also investigated this possibility. And in fact, a partial reduction of the thickening was observed in mice with existing cardiac hypertrophy in which RhoGEF12 was switched off. “We believe that RhoGEF12 is so important for the hypertrophy reaction because it combines signals from stretch and hormone receptors,” said Wettschureck.
The aim now is to develop a specific therapeutic process based on the insights gained from the study. Wettschureck’s group is thus currently investigating the question as to whether the molecular correlations discovered in the study are completely transferable to humans. Should this be confirmed, the next step should lead to the clinical application of the findings. Wettschureck is optimistic: “Two inhibitors have recently become known, which present possible candidates for a therapy. They could provide a basis for a pharmacological approach.” Another observation made by the study should also prove helpful in terms of developing a new therapeutic approach: the switching off of RhoGEF12 had no side effects in healthy mice.
MH/HR