Forschungsbericht 2014 - Max-Planck-Institut für molekulare Genetik
Molekulare Netzwerke in der Genom- und Proteomanalyse
Große Datensätze und molekulare Netzwerke
 wird in das Proteom (B, C, D) übersetzt. Dabei können genetische Veränderungen zu veränderten Proteinen führen, in diesem Beispiel zu drei Austauschen an je einer Aminosäure (B). Je nach Entwicklungsstadium, Zelltyp und individuellen Unterschieden werden wiederum die Proteine in unterschiedlichen Mengen produziert (C). Vor allem als Antwort auf Umwelteinflüsse oder umgebende Zellen können Proteine außerdem noch unterschiedlich post-translational modifiziert werden (D). Das Proteom ist somit um ein Vielfaches komplexer als das Genom. Unterschiede oder Veränderungen im Krankheitsfall können proteomumfassend festgestellt werden und beschreiben tausende molekulare Veränderungen der Zellen. Wichtige und nebensächliche Unterschiede sind jedoch aus diesen Daten nicht unmittelbar ersichtlich.")

Abb. 1: Ein Genom (A) wird in das Proteom (B, C, D) übersetzt. Dabei können genetische Veränderungen zu veränderten Proteinen führen, in diesem Beispiel zu drei Austauschen an je einer Aminosäure (B). Je nach Entwicklungsstadium, Zelltyp und individuellen Unterschieden werden wiederum die Proteine in unterschiedlichen Mengen produziert (C). Vor allem als Antwort auf Umwelteinflüsse oder umgebende Zellen können Proteine außerdem noch unterschiedlich post-translational modifiziert werden (D). Das Proteom ist somit um ein Vielfaches komplexer als das Genom. Unterschiede oder Veränderungen im Krankheitsfall können proteomumfassend festgestellt werden und beschreiben tausende molekulare Veränderungen der Zellen. Wichtige und nebensächliche Unterschiede sind jedoch aus diesen Daten nicht unmittelbar ersichtlich.
Technologische Weiterentwicklungen in den Bereichen DNA-Sequenzierung und Massenspektrometrie ermöglichen heute die Erstellung umfassender Datensätze, die Informationen über fast alle Moleküle der Zellen enthalten. Auf molekularer Ebene gibt es sehr viele Unterschiede zwischen unterschiedlichen Zelltypen, zwischen Zellen zu unterschiedlichen Zeitpunkten und zwischen gesunden und durch Krankheit veränderten Zellen. In der Krebsforschung werden durch intensive Sequenzierung der Genome einzelner Tumore 10.000 und mehr Variationen, zum Beispiel Austausche einzelner Nukleotidbausteine der DNA, die zu veränderten Proteinen führen, im Vergleich zum Genom des gesunden Gewebes festgestellt. Mit massenspektrometrischen Methoden können wiederum Veränderungen der Proteine, beispielsweise in fettleibigen Mäusen im Vergleich zu Mäusen mit Normalgewicht, genau bestimmt werden. In Krebszellen, deren Wachstumsprogamm nicht mehr ordnungsgemäß funktioniert, können an bestimmten Proteinen hunderte bis tausende posttranslationale Veränderungen, also Proteinmodifizierungen wie zum Beispiel Phosphorylierungen, gemessen werden, die in gesunden Zellen gar nicht oder an anderen Stellen vorkommen. Solche Unterschiede zwischen kranken und gesunden Zellen sind - mit entsprechendem Aufwand - gut messbar (Abb. 1). Dies stellt für die molekulare Medizin einen großen Fortschritt dar, weil zum ersten Mal auch komplizierte Veränderungen der Zelle gut und umfassend beschrieben werden können. Viele aus solchen Daten gewonnene Erkenntnisse tragen unmittelbar zum besseren Verständnis von krankhaften Veränderungen bei.
Bei solchen Messungen entstehen sehr große Datenmengen, was die Unterscheidung zwischen ursächlichen und nebensächlichen Veränderungen enorm erschwert. Das liegt auch daran, dass genom- und proteomweite Messungen an sich keine Informationen darüber geben, ob und wie die einzelnen Veränderungen der Moleküle miteinander zusammenhängen. In der Zelle aber wirken Proteine, RNAs, DNAs, Metabolite etc. niemals alleine, sondern kooperieren und wechselwirken in kleinen, mittleren und großen Gruppen miteinander. Die formalen Beschreibungen dieser Wechselwirkungen und Gruppen werden als molekulare Netzwerke bezeichnet. Sie stellen das sehr komplexe Zusammenspiel von tausenden unterschiedlicher Proteine, die Funktion und Aussehen einer Zelle bestimmen, sehr grundlegend dar. Die zu untersuchenden Moleküle, beispielsweise Proteine, werden als Knoten und deren Zusammenspiel als Kanten des Netzwerkes abgebildet, sodass sich dann die Struktur und Eigenschaften des Netzwerkes als Abbild der komplexen Zusammenhänge in der Zelle mathematisch beschreiben lassen (Abb. 2).
 ebenso wie zentrale Proteine (hubs) und Proteine an der Peripherie mathematisch erkennen.")
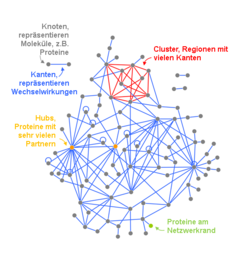
Bestimmte Strukturen sind universell und über die Zelle hinaus in fast allen natürlich entstandenen Netzwerken zu finden, zum Beispiel in neuronalen Netzwerken, in Ökosystemen, in sozialen Systemen oder auch in der Wirtschaft. Solche Netzwerkeigenschaften, die sich prinzipiell erst durch die systematische Messung möglichst großer Datensätze wie zum Beispiel alle Proteine oder Gene einer Zelle erschließen, könnten als Charakteristika komplexer Systeme universell sein [1]. In der Molekularbiologie ermöglicht die Erforschung von Netzwerken ein besseres Verständnis dafür, wie sich Zellen ihrem genetischen Programm folgend entwickeln und gleichzeitig durch Umwelteinflüsse verändert werden, das heißt, wie es zum Aussehen einer Zelle und ihren Funktionen kommt. Daneben bilden die manifestierten Netzwerke eine gute Basis für ein besseres Erkennen der ursächlichen Veränderungen in den beschriebenen großen Datensätzen, seien es genetische Variationen, Veränderungen der Proteinmengen oder Veränderungen der Proteine selbst durch Modifizierungen, weil sie die Zusammenhänge der wichtigen Veränderungen herstellen können.
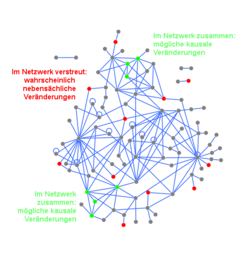
Bei komplexen Erkrankungen wie Krebs gibt es Veränderungen in einer Vielzahl von Genen, von denen aber nur einige zur Krankheitsentstehung oder deren Verlauf beitragen. Das Erkennen der relevanten molekularen Krankheitsveränderungen ist jedoch essenziell für ein grundlegendes Verständnis der Funktion der Zelle, genauso wie für die Diagnose und zielgerichtete Therapie einer Erkrankung. Bestimmte Veränderungen kommen im Genom, Proteom oder Phosphoproteom von Krebspatienten viel häufiger vor als andere, jedoch gibt es kein einzelnes Gen oder Protein, das allein für die Entstehung von Krebs verantwortlich ist. Ein typischer Krebspatient weist Veränderungen in ungefähr einem Dutzend von etwa 500 relevanten Genen auf. Obwohl die Art des Tumors in der Regel medizinisch klar diagnostiziert werden kann, scheint dennoch jeder Tumor jedes einzelnen Patienten eine Art molekulares Individuum zu sein [2]. Jedes Mutationsmuster eines Tumors ist zwar einzigartig, wichtige Veränderungen aber liegen in molekularen Netzwerken interessanterweise näher zusammen. Sie befinden sich in Bereichen mit vielen Kanten (cluster), da die betreffenden Moleküle in veränderten zellulären Prozessen zusammenwirken. Mit anderen Worten, funktionell wichtige Veränderungen sind innerhalb von Netzwerken in bestimmten Gruppen zu finden und können dadurch wesentlich besser von den vielen nebensächlichen Veränderungen unterschieden werden, die verstreut im Netzwerk lokalisiert sind (Abb. 3).

Erstellung und Analyse molekularer Netzwerke
Die Max-Planck-Forschungsgruppe Molekulare Interaktionsnetzwerke führt in ihrem Labor grundlegende Arbeiten zur Erstellung und Analyse von Protein-Protein-Wechselwirkungsnetzwerken beim Menschen durch. Die Forscher untersuchen systematisch, welche Proteine in der Zelle zusammenarbeiten - also temporär physikalisch miteinander verbunden sind - und in welchen zellulären Abläufen Gruppen von Proteinen zusammenwirken. Sie verwenden dafür ein genetisches Verfahren, dessen Prinzip vor 25 Jahren erstmalig beschrieben wurde: das Hefe-Zwei-Hybrid System [3]. Bei dieser Methode werden immer zwei Proteine in einem Hefestamm produziert, der genetisch derart verändert ist, dass ihm bestimmte Nährstoffe fehlen, die zum Überleben der Hefezellen wiederum dem Nährmedium zugemischt werden müssen. Erst wenn die beiden zu untersuchenden Proteine innerhalb der Hefezellen miteinander wechselwirken, können sie die betreffenden Nährstoffe selbst produzieren. Wächst diese Hefe im Experiment also ohne die Zusätze im Nährmedium, ist das ein deutlicher Hinweis darauf, dass die beiden Proteine miteinander interagieren. In zahlreichen Parallelversuchen mithilfe von Robotern und unter Anwendung neuer Sequenzierungsmethoden der zweiten Generation untersuchen die Wissenschaftler inzwischen sehr effizient, welche Proteine wechselwirken und erstellen im Anschluss Netzwerkkarten aus ihren Ergebnissen [4].
Für das Verständnis zellulärer Prozesse ist es von besonderem Interesse, welche Verbindungen zwischen Proteinen bestehen und welche dieser Verbindungen bei veränderten Bedingungen gelöst oder neu gebildet werden. Untersucht wird, welche Auswirkungen Veränderungen eines oder mehrerer Proteine auf ein Netzwerk haben und wie stark sich molekulare Netzwerke zwischen Individuen beziehungsweise zwischen Tumoren und gesundem Gewebe desselben Individuums voneinander unterscheiden. Mit experimentellen und computergestützten Ansätzen wollen die Forscher herausfinden, wie Proteinmodifizierungen Protein-Protein-Netzwerke beeinflussen und wie Zellen Proteinmodifizierungen "lesen" können [5].
Molekulare Netzwerke in der zellulären Signalverarbeitung
Zelluläre Signale können zum Beispiel durch Wachstumsfaktoren initiiert und anschließend durch posttranslationale Modifizierungen vorhandener Proteine innerhalb der Zelle weitervermittelt werden. Dabei häufig ist die Aktivierung sogenannter Kinasen an der äußeren Membran der Zelle, die daraufhin andere Proteine durch das Anhängen von Phosphatgruppen modifizieren. Als Folge werden hunderte Proteine in der Zelle phosphoryliert. Interessanterweise finden sich diese posttranslational modifizierten Proteine auch in wechselwirkenden Gruppen in Proteininteraktionsnetzwerken wieder [6]. Mit einer neuen Variante des Hefe-Zwei-Hybrid-Verfahrens haben Stelzl und sein Team begonnen, Wechselwirkungen zu untersuchen, die solche Phospho-Signale benötigen [7]. Das ist unter anderem deswegen interessant, weil Kinasen bei vielen Krebsarten aufgrund entsprechender Mutationen deutlich aktiver als im gesunden Organismus sind und dadurch zu mehr Zellteilungen und damit Tumoren führen.
Die Wissenschaftlerinnen und Wissenschaftler bestimmen systematisch nun alle diejenigen Wechselwirkungen, die durch erhöhte Kinaseaktivität in Krebszellen möglicherweise anders als im gesunden Gewebe verlaufen. So haben sie zum Beispiel eine Wechselwirkung genauer analysieren können, die erst durch Phosphorylierung auftritt und die in einfachen Zellkulturmodellen stärkere Zellmigration unterstützt. Verstärkte Zellmigration ist eine der charakteristischen Eigenschaften von Krebszellen. Diese Daten sind vor allem hilfreich, um besser zu verstehen, wie modifizierte Proteine ihre Eigenschaften ändern, wie diese Modifizierungen von ihren Wechselwirkungspartnern erkannt werden und wie in der Zelle Signale durch An- und Ausschalten von Proteinwechselwirkungen verarbeitet werden können.
Obwohl die hier vorgestellten Entwicklungen bereits vielversprechend sind, gibt es bedauerlicherweise jedoch noch keine Technik, mit der umfassend das Vorhandensein, die Abwesenheit oder die Stärke der Wechselwirkungen aller Proteine einer bestimmten Zelle oder eines Patienten parallel gemessen werden können. Die Forscher arbeiten daher jetzt daran, in sehr aufwendigen Experimenten eine Masterkarte aller möglichen Wechselwirkungen zu erstellen, die dann für die Interpretation von Daten aus spezifischen Proben, beispielsweise individueller Patienten, genutzt werden kann [8]. Die Analyse genom- und proteomweiter Messungen mithilfe von verschiedenen Referenznetzwerken zeigt nämlich schon heute, dass für viele Prozesse die quantitative Messung lediglich einer begrenzten Anzahl von Molekülen schon ausreichen könnte, um molekulare Prozesse insgesamt besser zu verstehen und einzuordnen, zelluläre Phänotypen besser vorherzusagen und Krankheiten besser diagnostizieren zu können.
Literaturhinweise
DOI: 10.1038/nphys2188
DOI: 10.1126/science.1235122
DOI: 10.1038/nmeth.3182
DOI: 10.1038/nmeth.2397
DOI: 10.1016/j.sbi.2013.11.009
DOI: 10.1371/journal.pcbi.1002933
Molecular Systems Biology 11: 794 (2015)
DOI: 10.15252/msb.20145968
DOI: 10.1002/prca.201300039