Forschungsbericht 2003 - Max-Planck-Institut für Polymerforschung
Polymertheorie: Von spezifischen Eigenschaften nahe einer Metallgrenzfläche bis zum Chromatin
Theorie der Polymere (Prof. Dr. Kurt Kremer)
MPI für Polymerforschung, Mainz
Ausgangspunkt der theoretischen Untersuchung ist die Tatsache, dass weiche Materie im Allgemeinen und Polymere im Speziellen von großen Fluktuationen dominiert werden. Anders formuliert heißt das, die charakteristische Energieskala ist die thermische Energie kBT und somit von gleicher Größenordnung wie die Entropie: Die Dichte der (freien) Energie ist um Größenordnungen niedriger als bei konventionellen Festkörpern. Das hat zur Folge, dass zum Verständnis viele unterschiedliche Gesichtspunkte beitragen, deren Einfluss mit zum Teil sehr unterschiedlichen theoretischen Methoden untersucht wird. Diese reichen von quantenchemischen Verfahren bis zur makroskopischen hydrodynamischen Theorie.
Beispielhaft sollen drei Projekte kurz vorgestellt werden, die sehr unterschiedliche Aspekte beleuchten.
Polymere mit spezifischen Wechselwirkungen an Grenzflächen
Die Wechselwirkung von Polymeren mit festen Grenzflächen wird theoretisch im Allgemeinen auf einem phänomenologischen Niveau diskutiert, das heißt die Wand, mit der die Wechselwirkung stattfinden kann, ist entweder repulsiv oder übt eine kurzreichweitige, attraktive Wechselwirkung auf die Monomere der Ketten aus. Derartige Ansätze geben ein gutes qualitatives Bild des Verhaltens von Makromolekülen an Grenzflächen, können allerdings spezifische Wechselwirkungen nicht berücksichtigen. Sie können auftreten, wenn einzelne Teile der Kette, Monomere oder Untereinheiten der Monomere, besonders stark von der Oberfläche abgestoßen bzw. angezogen werden. Um das besser zu verstehen, ist eine explizite Berücksichtigung der Details der chemischen Natur der Ketten in Verbindung mit den globalen Konformations-Eigenschaften notwendig. Über die letzten Jahre haben wir uns im Rahmen der Entwicklung von Multiskalensimulationsverfahren mit genau diesen Fragen intensiv auseinander gesetzt. Als zentrales Beispiel für die Entwicklung und Anwendung einer solchen Methodik diente die Adsorption von Bisphenol-A-Polykarbonat (BPA-PC) auf einer Nickeloberfläche. Dieses System weist einerseits eine hohe Komplexität, also eine Herausforderung für die Theorie, auf und ist andererseits von nicht zu unterschätzendem technischem Interesse. Die Vorgehensweise ist dabei in Abbildung 1 illustriert.
)")
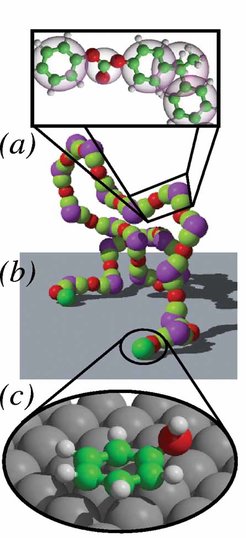
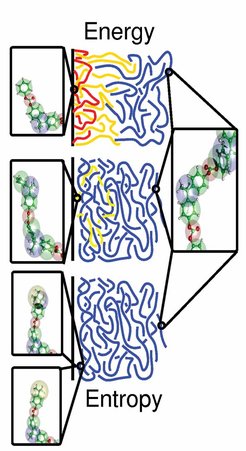
Zunächst wurde die Wechselwirkung von Fragmenten der Wiederholungseinheit des Polycarbonats (Karboxylsäure, Propan, Benzol und Phenol) mit quantenmechanischen Rechnungen (CPMD) im Detail untersucht. Dabei stellte sich heraus, dass der Benzolring sehr stark von der Nickeloberfläche angezogen wird, diese Anziehungskraft allerdings nur kurzreichweitig ist. Andererseits erfährt die Karboxylsäure bzw. das Propanmolekül eine sehr starke, längerreichweitige, ausschließlich abstoßende Wechselwirkung mit der Metallgrenzfläche. Diese schirmt die Adsorption der Benzolringe ab. Die gesamte Oberfläche wirkt für die Kette zunächst einmal repulsiv. Ganz anders ist es bei den phenolartigen Kettenenden, die in sehr vielen Fällen auf Grund des Polymerisationsprozesses vorhanden sind. Hier findet diese Abschirmung durch die anderen Konstituenten des Monomers nicht statt, sodass die Kettenenden stark (bei Raumtemperatur mit etwa 40 kBT) an der Oberfläche chemisorbieren können. Diese Information wurde dann benutzt, um ein systematisch entwickeltes vergröbertes Modell von Polykarbonat, das die intrinsischen Abhängigkeiten der Bindungswinkel und Bindungslängen statistisch richtig berücksichtigt, zu parametrisieren. Damit war es möglich, eine solche Schmelze in der Nähe einer Metallgrenzfläche realitätsnah mit einer Simulation zu untersuchen. In einer systematischen Studie mit anderen Kettenenden konnte gezeigt werden, dass das ganze Spektrum von Kettenmorphologien an einer Grenzfläche von so genannten "sticky ends" bis hin zu einer insgesamt neutralen repulsiven Grenzfläche durch Variation der Kettenstopper in der Polymerisation abgedeckt werden kann. Abbildung 2 fasst diese Ergebnisse für die Schmelzenmorphologie als Funktion der unterschiedlichen Kettenenden zusammen. Um ein solches Ergebnis zu erzielen, ist es unabdingbar, die verschiedenen Skalen der Simulation, nämlich die quantenmechanische lokale Wechselwirkungsrechnung, eine atomistische Modellierung lokaler Bindungswinkel und Torsionswinkel sowie die vergröberte Modellierung der Schmelze in der Nähe der Oberfläche miteinander zu verbinden. Zurzeit werden diese Ansätze auf andere Systeme bzw. andere Grenz- und Oberflächen übertragen. Dabei kann man sowohl an eine gezielte Aggregation von Molekülen denken als auch an Ordnung, die z. B. durch Defektlinien in der Oberfläche in der Polymerschmelze induziert werden (Delle Site, van der Vegt, Kremer).
-Oberfläche für die unterschiedlichen Kettenenden. Die Morphologien variieren von Adsorptionsenergie-dominierten \"sticky ends\" bis zu Packungsentropie-dominierten Konformationen nahe einer harten repulsiven Wand (L. Delle Site, S. Leon, K. Kremer, Journal of the American Chemical Society 126, 2944-2955 (2004)).")
Kettenkonformation und Modul von Schmelzen
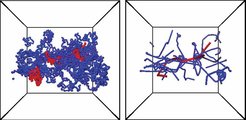
Polymere Schmelzen zeichnen sich durch ein faszinierendes visko-elastisches Verhalten aus, das heißt, auf langen Zeitskalen verhalten sie sich wie Flüssigkeiten, während sie sich auf kurzen Zeitskalen, die für sehr lange Ketten durchaus Minuten oder gar Stunden sein können, wie weiche elastische Festkörper verhalten. Das Verständnis dieses Phänomens und die quantitative Vorhersage der elastischen Module ist seit jeher ein zentrales Feld der theoretischen und experimentellen Polymerforschung. Die Ursache für dieses festkörperartige Verhalten in dem intermediären Zeitbereich wird üblicherweise im Rahmen des Röhrenkonzeptes von Edwards und DeGennes diskutiert. Diese Röhre ist keine feste Röhre, in der sich die Kette befindet, sondern eine Veranschaulichung der Bewegungseinschränkung der Kette durch all die anderen Polymere in ihrer Umgebung. Die für jedes Polymer charakteristische Länge ist dabei der so genannte Röhrendurchmesser oder äquivalent typische Zahl von Kettenmonomeren, die im Kettenknäuel in der Röhre vorliegen. Betrachtet man diese Größe als anpassbaren Parameter, d. h. als Messgröße bzw. Fitgröße, so kann man mit den gängigen Theorien das Verhalten vieler Systeme recht gut beschreiben. Was bisher fehlte und seit vielen Jahren intensiv diskutiert wird, ist eine Möglichkeit diese charakteristische Länge aus den Konformationen der Ketten vorherzusagen und damit auch Vorhersagen für das Verhalten bestimmter Materialien zu machen. Die vielfach untersuchten Ansätze im Rahmen der Knotentheorie, d. h. einer Analyse der Verschlaufungen von zwei benachbarten Polymerfäden, können dies nicht leisten, da die charakteristischen Vielketteneffekte nicht ausreichend berücksichtigt werden können. In diesem Zusammenhang wurde ein alternativer Ansatz entwickelt, der auf das ursprüngliche Konzept des primitiven Pfades von Edwards zurückgeht. Der entscheidende Unterschied ist dabei, dass zur Konstruktion dieses primitiven Pfades, den man als kürzesten Weg zwischen den Endpunkten der Kette, der keine anderen Ketten durchschneidet, beschreiben kann, gleichzeitig in konsistenter Weise sämtliche Ketten berücksichtigen muss. Daraus kann man das Gerüst der primitiven Pfade einer Schmelze bestimmen und Röhrendurchmesser ableiten. Abbildung 3 zeigt ein typisches Beispiel der Kettenkonformation vor und nach dieser Prozedur.
.")
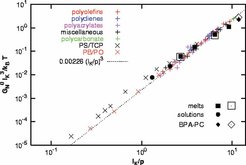
Setzt man diese Ergebnisse in die Theorie für den elastischen Modul von Doi und Edwards ein und trägt die Daten in geeignet skalierter Form auf (Um Simulation und Experiment vergleichen zu können, muss auf dimensionslose Größen übergegangen werden. Die geeigneten Längenskalierungen ergeben sich aus Steifheit und der Packungsdichte der Kette), so findet man, dass man mit diesem Ansatz sowohl Lösungen als auch Schmelzen (semi)-quantitativ beschreiben kann. Die Daten überstreichen einen Bereich von über sechs Dekaden im Modul. Innerhalb der charakteristischen Messgenauigkeit stimmen sie hervorragend mit den Experimenten für sehr viele völlig unterschiedliche Polymere überein. Damit ist erstmals ein direkter Zugang zu dieser Größe aus den Konformationen der Kette heraus gewonnen worden. Zurzeit wird dieser Ansatz auf verschiedene Systeme erweitert, insbesondere auch zu semi-flexiblen Ketten hin. Abbildung 4 fasst das Ergebnis des Vergleiches von Simulation und Experiment zusammen.
 (R. Everaers et al., Science 303, 823-826 (2004)).")
In ähnlicher Form, nur mit analytischer Theorie und nicht mit Simulationsverfahren beschreibt das letzte Beispiel, wie man mit sehr vereinfachten Modellen zu durchaus für die Experimente relevanten Informationen kommen kann.
Polymertheorie und Biopolymere
Theoretische Untersuchungen zu Biopolymeren kann man im Allgemeinen in zwei Gruppen einteilen. Einerseits werden in atomistischen Simulationen lokale Details, insbesondere im Hinblick auf ihre Funktion hin, untersucht. Andererseits werden mit sehr vergröberten generischen Modellen allgemeine Aspekte der Struktur und Dynamik von Biomolekülen diskutiert. Ein solches Beispiel sind die physikalischen Grundlagen für die Möglichkeit der DNS entlang der Histonkomplexe (Octamere aus Histonproteinen), um die sie gewickelt ist, zu diffundieren oder zu gleiten. Dies geschieht, obwohl die DNS lokal sehr stark an diese Proteine gebunden ist. Diese Mobilität der DNS ist von entscheidender biologischer Bedeutung, da die Transkription nur möglich ist, wenn das entsprechende DNS-Stück nicht fest an das Histon-Oktamer gebunden ist. Für die Bewegung dieser Nukleosomen entlang der DNS, ausgehend von spontanen thermischen Fluktuationen, kann man zwei Szenarios entwickeln, die beide auf der Aktivierung von Zwischenzuständen beruhen: Einmal die Repositionierung über Schleifen- und zum Anderen über Torsionsdefekte der DNS. Geht man von den Standardparametern, nämlich der elektrostatischen Anbindung und der bekannten Torsion- bzw. Biegesteifheit der DNS aus, sind zunächst beide Mechanismen plausibel. Die typischen Energien liegen in beiden Fällen in dem Bereich, der durch thermische Fluktuationen erreichbar ist. In einer detaillierten Analyse der Experimente im Vergleich zu analytischen Theorien konnte nun gezeigt werden, dass typische In-vitro-Experimente, bei denen synthetische Liganden Torsionsdefekte blockierten und somit nur noch Schleifendefekte zuließen, dazu führten, dass die Mobilität komplett unterdrückt war, d. h. Torsionsdefekte sind für die thermische Anregung der Diffusion entlang der DNS notwendig. Andererseits stehen dem In- vivo-Experimente gegenüber, die zeigen, dass über ATP getriebene molekulare Motoren die DNS offensichtlich entlang der Nukleosome schieben oder ziehen können. Hier geht man davon aus, dass dies durch induzierte Schleifendefekte aber nicht durch Torsionsdefekte ermöglicht wird. Das heißt, zurzeit ergibt sich aus Experiment und Theorie, dass Torsionsdefekte für die fluktuationsinduzierte Bewegung der Nukleosomen entlang der DNS verantwortlich sind, während Schleifendefekte wahrscheinlich über ATP getriebene molekulare Maschinen erzeugt werden können. Abbildung 5 illustriert die Mechanismen, wie sie sich nach der jetzigen Erkenntnis in der Diskussion zwischen Theorie und Experiment darstellen (Schiessel).
 illustriert (I. Kulic, H. Schiessel, Biophysical Journal 84, 3197-3211 (2003)).")
