Forschungsbericht 2004 - Max-Planck-Institut für Biochemie
Hochauflösende Mikroskopie von Zellen und Oberflächen: Kryo-Elektronentomographie und Raster-Infrarotmikroskopie im optischen Nahfeld
Molekulare Strukturbiologie (Baumeister) (Prof. Dr. Wolfgang Baumeister)
MPI für Biochemie, Martinsried
Kryo-Elektronentomographie
Die dreidimensionale (3D) Struktur und funktionelle Organisation von Viren, Prokaryoten und Eukaryotenzellen im Nanometerbereich (millionstel Millimeter) ist uns auch heute noch weitgehend unbekannt. Mit der Entwicklung der Kryo-Elektronentomographie bietet sich erstmals die Möglichkeit, die 3D-Struktur biologischer Zellen bei molekularer Auflösung zu untersuchen. Das grundsätzliche Konzept hierfür war nicht neu, doch es sind wesentliche methodische und technische Fortschritte in der Elektronenmikroskopie und Bildverarbeitung notwendig gewesen, um diese Technik erfolgreich umsetzen zu können. Unsere Abteilung hat die automatisierte Kryo-Elektronentomographie für die Anwendung an strahlenempfindlichen biologischen Objekten entwickelt und arbeitet an ihrer weiteren Verbesserung.
Die Methode der Kryo-Elektronentomographie
Die funktionelle Struktur biologischer Zellen bleibt in einem nahezu nativen Zustand erhalten, wenn die Objekte bei –190 °C schockgefroren und ohne chemische Fixierung und Kontrastierung mikroskopiert werden. Will man in amorphes Eis eingebettete Zellen bei molekularer Auflösung dreidimensional rekonstruieren, so steht man vor dem Dilemma, zwei sich widersprechende Anforderungen zu erfüllen: Einerseits werden möglichst viele, aus unterschiedlichen Richtungen abgebildete Projektionen des Objekts benötigt, um die räumliche Struktur gut aufgelöst rekonstruieren zu können; andererseits sind biologische Proben so strahlenempfindlich, dass die Elektronendosis begrenzt werden muss. In den letzten Jahren wurden die technischen Voraussetzungen geschaffen und Methoden entwickelt, um aus einer Serie von Niederdosis-Aufnahmen (Kippserie) 3D-Datensätze intakter Zellen zu gewinnen, welche strukturelle Information bis zu einer Auflösung von derzeit 4 – 6 nm enthalten. Der entscheidende Fortschritt bestand in der Automatisierung der Bildaufzeichnung und der Steuerung des Elektronenmikroskops. Dadurch war es möglich, fast die gesamte Strahlendosis für die Aufzeichnung der Projektionen zu nutzen. Datenakquisition, Aufbereitung der Aufnahmen, 3D-Rekonstruktion und Bildanalysefunktionen wurden in dem Programm TOM software toolbox zusammengefasst [1].
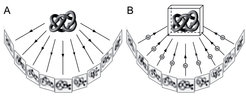
Das Prinzip der Elektronentomographie ist es, ein Objekt im Elektronenmikroskop um eine Achse zu kippen und von einem großen Winkelbereich Projektionen aufzuzeichnen (Abb. 1). Da der dünne Eisfilm und die darin enthaltenen Zellen von einem stabilen Objektträger gehalten werden, ist es aber nicht möglich, Aufnahmen bis zu einem Kippwinkel von +/- 90° zu gewinnen. Als Folge des für die 3D-Rekonstruktion nur unvollständigen Datensatzes erscheinen in den Tomogrammen manche Strukturen, abhängig von ihrer Orientierung, mit schwachem Kontrast und reduzierter Auflösung. Um diese Effekte zu mildern, wurde ein Dreh-Kipphalter entwickelt, der es erlaubt, die Probe bei niedrigen Temperaturen im Mikroskop um ihre z-Achse zu drehen. So kann eine zweite Kippserie des gleichen Objekts mit anderer Orientierung aufgenommen und mit der ersten Serie kombiniert werden. Dadurch wird der Bereich fehlender 3D-Information deutlich verringert.
 Durchstrahlung eines dreidimensionalen Objekts unter verschiedenen Winkeln im Elektronenmikroskop und Aufnahme einer Serie von zweidimensionalen Projektionen (Kippserie). (B) Rückprojektion der im Computer gespeicherten Bilddaten in ein gemeinsames Volumen mit 3D-Rekonstruktion des abgebildeten Objekts. Die Projektionsdaten werden zur Rekonstruktion mit einer geeigneten Funktion (W) gewichtet.")
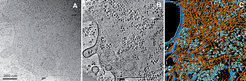
Die Rekonstruktion erfolgt durch das Prinzip der gefilterten Rückprojektion, bei der alle Aufnahmen im Computer zueinander ausgerichtet und in einem umgekehrten Projektionsprozess in ein gemeinsames Volumen eingefügt werden. Niederdosisaufnahmen von nativen, in Eis eingebetteten Zellen weisen jedoch ein sehr niedriges Signal-zu-Rausch-Verhältnis auf und stellen damit die Verarbeitung der Bilddaten vor das Problem, Rauschbeiträge von Strukturdetails verlässlich zu trennen und aus dem Tomogramm zu entfernen. Hierfür mussten erst neue Ansätze gefunden werden (nichtlineare anisotrope Diffusion), da herkömmliche Filterverfahren nicht zum Ziel führten. Eine weitere Herausforderung liegt darin, verschiedene, aber oft eng benachbarte Strukturen in Tomogrammen voneinander abzugrenzen (Segmentierung). Ein erfolgreiches Verfahren weist jedem dreidimensionalen Volumenelement eine fraktale Dimension zu, die ein Indikator für die Zugehörigkeit des Bildpunktes zu einer zusammenhängenden Struktur (Elektronendichte) darstellt. Auf diese Weise gelang es zum Beispiel, Membranen, das Cytoskelett und große makromolekulare Komplexe in Tomogrammen des Schleimpilzes Dictyostelium discoideum zu identifizieren und zu visualisieren (Abb. 2). So konnte erstmals die Anordnung und das Verzweigungsmuster der Aktinfilamente in intakten Zellen untersucht und das Aktinnetzwerk in verschiedenen Funktionszuständen von Filopodien der amöboid beweglichen Zellen darstellt werden [2]. Mithilfe der Kryo-Elektronentomographie zeigte sich auch, dass das Cytoskelett des in Bienen lebenden Bakteriums Spiroplasma melliferum wesentlich komplexer strukturiert ist als bisher vermutet. Es besteht aus mehreren, parallel verlaufenden Bändern unterschiedlich dicker Filamente. Diese besondere Anordnung bildet die Grundlage für ein Modell, mit dem sich die außergewöhnliche, Flagellen-unabhängige Bewegung des spirillenförmigen Bakteriums erklären lässt [3].
 Projektionsbild der in amorphes Eis eingebetteten, unbehandelten Zelle. Das Bild ist kontrastarm und durch einander überlagernde Strukturen schwer interpretierbar. (B) Eine isoliert dargestellte dünne Schicht des 3D-Volumens der rekonstruierten Zelle. (C) Visualisierung der 3D-Struktur nach Segmentierung. Die Aktinfilamente des Cytoskeletts sind orange, große Proteinkomplexe (Ribosomen) hellblau und Lipidmembranen blau dargestellt.")
Der Kernporenkomplex von Dictyostelium discoideum
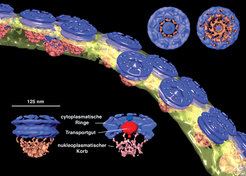
Die Hülle des Kerns eukaryotischer Zellen enthält zahlreiche Poren, die von einem komplizierten Kernporenkomplex gebildet werden. Mindestens 30 verschiedene Proteine bauen den oktameren Komplex auf, der den nucleo-cytoplasmatischen Stoffaustausch von Ribonukleinsäuren und Proteinen kontrolliert. Tomogramme Transport-aktiver Zellkerne von D. discoideum enthalten die Struktur einer ganzen Reihe solcher Komplexe. Extrahiert man die entsprechenden Subvolumina der Tomogramme im Computer, so können sie klassifiziert und anschließend 3D-gemittelt werden. Abbildung 3 zeigt ein Modell des Kernporenkomplexes bei einer isotropen Auflösung von etwa 8 nm. Die strukturelle Klassifizierung zeigt, dass die zentrale Masse – das durch den Porenkomplex transportierte molekulare Gut – in unterschiedlichen Positionen erscheint und sich die Konformation des Gesamtkomplexes verändert [4]. Mithilfe von Kryo-Elektronentomographie lassen sich also verschiedene funktionelle Zustände von Molekülkomplexen in vivo erfassen.
, die filigrane, korbähnliche Struktur liegt im Kernplasma (Aufsicht rechts oben). Der Porenkomplex ist in die Doppelmembran der Kernhülle (gelb) eingebettet. Die rot und teils blau markierte Masse in der Mitte des aufgeschnitten dargestellten Porenkomplexes entspricht dem molekularen Transportgut, das verschiedene Positionen in den rekonstruierten Komplexen einnimmt.")
Kernporenkomplexe sind groß und ihre Konformation ist so heterogen, dass sich die Struktur kaum durch Röntgenstrukturanalyse als Ganzes aufklären lassen wird. Sind aber die atomaren Strukturen der einzelnen, isolierten Komponenten erst einmal bekannt, bietet die Elektronentomographie die Möglichkeit, sie in ein Tomogramm des Gesamtkomplexes einzupassen und ein pseudoatomares Modell zu erstellen. Diese Hybridtechnik eröffnet einen Weg, strukturelle Informationen mit unterschiedlichen Auflösungsniveaus zu integrieren [5].
Kartierung makromolekularer Komplexe
Die Kryo-Elektronentomographie bildet als nicht-invasive Technik Zellen in einem lebensnahen Zustand ab. Tomogramme liefern im Grunde 3D-Datensätze des gesamten Proteoms bei molekularer Auflösung und erlauben damit Einblicke in die Zusammensetzung, Verteilung und Interaktion makromolekularer Komplexe in einer individuellen Zelle.
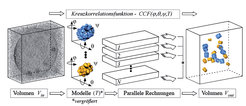
Da Kryo-Elektronentomogramme ein niedriges Signal-zu-Rausch-Verhältnis aufweisen und das Cytoplasma eine hohe Konzentration an Proteinen in dichter Anordnung enthält, stößt die Technik der Segmentierung schnell an Grenzen. Es musste deshalb ein neuer Weg zur Lokalisierung und Identifizierung makromolekularer Komplexe beschritten werden. Mithilfe von 3D-Korrelation wird nun die strukturelle Signatur eines Komplexes mit bekannter Struktur in dem gesamten Tomogramm einer Zelle gesucht (template matching). Dieses Verfahren ist sehr rechenintensiv: Das Zielmolekül muss in allen Orientierungen mit dem gesamten 3D-Datensatz der Zelle korreliert werden, um es verlässlich identifizieren zu können (Abb. 4). Dass dieser Ansatz prinzipiell gangbar ist, zeigten erste Rechnungen mit so genannten „Phantomzellen“. Dazu wurden Lipidvesikel mit zwei verschiedenen, aber ähnlich großen Proteinkomplexen (Proteasomen und Thermosomen) gefüllt, mittels Kryo-Elektronentomographie 3D-rekonstruiert und die Moleküle über das Korrelationsverfahren detektiert [6].
 im Tomogramm einer 3D-rekonstruierten intakten Zelle (links). Die Datenvolumina der Zelle und des Modellmoleküls werden in allen möglichen räumlichen Orientierungen kreuzkorreliert. Das Verfahren ist rechenaufwändig und erfolgt in Parallelrechnern. Das Ergebnis ist eine dreidimensionale Korrelationsfunktion, die die Position und Orientierung der im Zellvolumen identifizierten Moleküle angibt. Die Molekülstrukturen können nun in ein Modellvolumen der Zelle eingefügt und ihre Verteilung untersucht werden (rechts).")
Inzwischen konnten auf diesem Weg Ribosomen in Tomogrammen des Bakteriums Spiroplasma melliferum lokalisiert werden (Abb. 5). Die Identifizierung auch kleinerer Proteinkomplexe wird möglich sein, wenn sich die Auflösung der Tomogramme auf etwa 2 nm steigern lässt. Dies erfordert weitere technische Verbesserungen und 3D-Datensätze mit nahezu isotroper Auflösung, die durch Zweiachsen-Kippung zu erreichen ist.
Das Proteom von Thermoplasma acidophilum – Zellulärer Atlas
Thermoplasma acidophilum ist ein frei lebendes, thermophiles Archaeon mit relativ kleinem Genom, das vollständig sequenziert wurde [7] und dessen Proteom in unserer Abteilung analysiert wird. Insbesondere interessieren uns Enzyme der Protein-Qualitätskontrolle (intrazelluläre Proteasen und Chaperone), die meist größere und im Falle der Tricorn-Protease sehr große Komplexe bilden (Abb. 5). Es ist das langfristige Ziel, alle identifizierbaren Komplexe in Thermoplasma-Zellen zu kartieren und ihre Interaktionen in vivo aufzuklären. Allerdings sind erst wenige Proteine strukturell bekannt. Deshalb soll ein Atlas der cytoplasmatischen Proteinkomplexe erstellt werden, um sie dann in Tomogrammen zuordnen zu können. Dazu werden die isolierbaren Komplexe mit Kryo-Elektronenmikroskopie abgebildet und mittels Einzelpartikelanalyse 3D-rekonstruiert. Um die Struktur möglichst vieler verschiedener Partikel bei guter Auflösung zu erhalten, ist ein hoher Durchsatz von Proben und Daten erforderlich. Einzelne Schritte der Präparation, Datenakquisition und Datenanalyse sollen dafür automatisiert werden.
Die Möglichkeit, das Interaktionsnetzwerk makromolekularer Komplexe im funktionellen Zustand einzelner Zellen abbilden und auswerten zu können, gehört zu den faszinierenden Perspektiven der Kryo-Elektronentomographie.
![(A) Rekonstruktion des Bakteriums Spiroplasma melliferum und Analyse der Verteilung und Orientierung von Ribosomen (gelb) mithilfe des 3D-Korrelationsverfahrens. (B) 3D-Modelle großer Proteinkomplexe. Links: Der einer Doppelspirale ähnelnde Superkomplex der Tripeptidyl-Peptidase II aus der Fruchtfliege Drosophila melanogaster [9]. Rechts: Das aus 20 Hexameren gebildete Kapsid der Tricorn-Protease aus dem Archaeon Thermoplasma acidophilum [8].](/817691/original-1294357518.jpg?t=eyJ3aWR0aCI6MzQxLCJmaWxlX2V4dGVuc2lvbiI6ImpwZyIsIm9ial9pZCI6ODE3NjkxfQ%3D%3D--e15a2e70531c51081a8efc689c29553f79d948f7 "(A) Rekonstruktion des Bakteriums Spiroplasma melliferum und Analyse der Verteilung und Orientierung von Ribosomen (gelb) mithilfe des 3D-Korrelationsverfahrens. (B) 3D-Modelle großer Proteinkomplexe. Links: Der einer Doppelspirale ähnelnde Superkomplex der Tripeptidyl-Peptidase II aus der Fruchtfliege Drosophila melanogaster [9]. Rechts: Das aus 20 Hexameren gebildete Kapsid der Tricorn-Protease aus dem Archaeon Thermoplasma acidophilum [8].")
![(A) Rekonstruktion des Bakteriums Spiroplasma melliferum und Analyse der Verteilung und Orientierung von Ribosomen (gelb) mithilfe des 3D-Korrelationsverfahrens. (B) 3D-Modelle großer Proteinkomplexe. Links: Der einer Doppelspirale ähnelnde Superkomplex der Tripeptidyl-Peptidase II aus der Fruchtfliege Drosophila melanogaster [9]. Rechts: Das aus 20 Hexameren gebildete Kapsid der Tricorn-Protease aus dem Archaeon Thermoplasma acidophilum [8].](/817691/original-1294357518.jpg?t=eyJ3aWR0aCI6MjQ2LCJvYmpfaWQiOjgxNzY5MX0%3D--f2df6afd2ae2e9d05e80f644285202ab3000aa70)
Raster-Infrarotmikroskopie im optischen Nahfeld
In der Lichtmikroskopie galt die Wellenlänge des Lichts als eine Auflösungsgrenze, die ganz grundsätzlich ein Erkennen kleinerer Strukturen ausschloss. In den vergangenen zwei Jahrzehnten aber haben optische Nahfeldmethoden diese Grenze aufgehoben und damit überraschend auch Licht sehr langer Wellenlängen für die Mikroskopie einsetzbar gemacht. Insbesondere ist der Wellenlängenbereich des mittleren Infrarots interessant, weil er die charakteristischen Schwingungen (molecular fingerprints) aller chemischen Substanzen umfasst. Infrarotspektrometer sind daher zur Identifizierung von chemischen Substanzen aus Forschungs- wie Industrielabors nicht mehr wegzudenken.
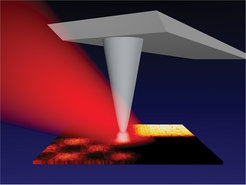
Unsere Entwicklung von Infrarot-Nahfeldmikroskopen basiert auf Rasterkraftmikroskopen, die für sich allein die Oberfläche – die Topographie – einer Probe mithilfe einer Abtastspitze abbilden können. Wird diese Spitze zusätzlich mit Infrarotstrahlung beleuchtet, ergibt sich durch einen Antenneneffekt genau am Spitzenapex eine erhöhte Infrarotintensität (Abb. 6). Dieser „Nahfeld-Fokus“ liefert die gewünschte punktuelle Information über die Infrarotabsorption, und zwar dadurch, dass das ausgestreute Infrarotlicht an einem Detektor aufgezeichnet wird. Das Erstaunliche dieses Messvorgangs ist, dass durch die Rasterung ein Infrarot-Absorptionsbild mit sehr guter Ortsauflösung entstehen kann, das allein durch den Spitzendurchmesser festgelegt wird und mit derjenigen des zugleich erhaltenen Topographiebilds übereinstimmen sollte [10, 11]. Dies bedeutet, dass mit handelsüblichen Tastspitzen, deren Durchmesser etwa 20 nm beträgt, Infrarotbilder mit einer lateralen Auflösung von ebenfalls 20 nm erhalten werden können, also dem Tausendstel einer zur Beleuchtung benutzten Wellenlänge von 20 µm. Unsere Experimente mit nanometrischen Objekten aus Halbleitern, Metallen und Dielektrika konnten diese Erwartung bestätigen. Ein wesentlicher technischer Kniff dabei war ein neuartiges elektronisches Filterverfahren, mit dessen Hilfe die hohen Störsignale unterdrückt werden konnten, die von der Infrarotstreuung in der Nähe der Spitze befindlicher Objekte, wie etwa der Hebelaufhängung, herrühren.
 einer Probe abbildet, erzeugt ein schräg einfallender, fokussierter Infrarotstrahl einen am Spitzenende konzentrierten, sehr hellen Beleuchtungsbereich. Dieser in der Praxis ca. 20 nm messende „Nahfeldfokus“ erzeugt durch Aufzeichnung des ausgestreuten Infrarotlichts ein nach klassischen Erwartungen unmöglich hoch aufgelöstes Infrarotbild (< λ/100) der Oberfläche, hier eines Testmusters.")
Damit sich die chemische Information in deutlichen Infrarotkontrasten ausdrückt, muss eine geeignete Wellenlänge der Beleuchtung ausgewählt werden. Das sehr breite, kontinuierliche Spektrum von thermischen Lichtquellen, wie sie in Fourier–Transform–Infrarot- Spektrometern verwendet werden, scheidet wegen der geringen Leistungsdichte für die Zwecke der Infrarot-Nahfeldmikroskopie aus. In der Anfangsphase unserer Forschung stand daher lediglich monochromatische Beleuchtung mit Wellenlängen eines begrenzten Gebiets von 9 – 11 µm zur Verfügung, die von sequenziell einstellbaren Emissionslinien eines CO2–Lasers herrührte. Kürzlich konnte durch einen CO–Laser das Wellenlängengebiet von 5 –7 µm hinzugenommen werden. Diese spektrale Erweiterung ermöglichte den Zugang zu deutlich ausgeprägten Infrarotresonanzen von organischen Materialien und Proteinen.
An der resonanten Schwingung des Kunststoffs Polymethylmethacrylat bei 5,8 µm wurden systematische und quantitative Studien zur Entstehung des Kontrasts in der Nahfeldmikroskopie durchgeführt [12]. Erstmals konnte theoretisch und experimentell gezeigt werden, dass die Helligkeitskontraste nicht einer spektralen Glockenkurve folgten (wie es bei Absorption der Fall wäre), sondern einer Dispersionskurve mit einem Maximum auf der niederfrequenten Flanke der Resonanz und einem Minimum auf der höherfrequenten Flanke. Überraschend und von hoher Bedeutung für die Anwendbarkeit der Methode ist, dass diese spektralen Nahfeldkontraste erst abnehmen, wenn die Materialtiefe geringer als etwa ein Spitzendurchmesser wird. Die Methode ist somit sehr empfindlich, da die vollen Kontraste bereits mit sehr geringer Materialdicke erreicht werden. In der klassischen Infrarotspektroskopie hingegen sind stets um Größenordnungen höhere Schichtdicken zur Messung einer Absorptionskurve nötig. Diese Erkenntnisse wurden an Proteinpräparaten bestätigt, die im Wellenlängenbereich um 6,1 µm eine ausgeprägte, von der Sekundärstruktur beeinflusste Schwingungsbande aufwiesen. Eine erste Untersuchung biologischer Strukturen gelang mit der Abbildung eines einzelnen Tabakmosaikvirus. Das Virus (Durchmesser 18 nm) wurde zwanzigmal nacheinander mit jeweils veränderter Wellenlänge im Bereich von 5,7 – 6,3 µm abgebildet. Jedem Bildpunkt konnte ein lokales Infrarotspektrum des Virusproteins zugeordnet werden, das den erwarteten dispersiven Verlauf mit gut ausgeprägtem Kontrast zeigte.
Sensitivität und Spezifität versprechen der Infrarot-Nahfeldmikroskopie ein interessantes Anwendungspotenzial in den Nanowissenschaften. Um dies zu realisieren, ist der Übergang auf eine parallel-polychromatische Beleuchtung und eine breite spektrale Überdeckung unumgänglich. Als ersten Schritt in diese Richtung haben wir kürzlich ein neuartiges, kohärentes Kamm-Spektrometer vorgestellt [13].