Forschungsbericht 2013 - Max-Planck-Institut für molekulare Physiologie
Die Ras-Abhängigkeit von Tumoren im Visier der Wissenschaft
Einleitung
Eine der häufigsten Krankheiten mit Todesfolge in Deutschland ist Krebs. Zu Beginn dieser Krankheit erlangen einzelne Zellen durch Mutationen in ihrem Erbgut, der DNS, einen Wachstumsvorteil gegenüber den Zellen in ihrer Umgebung. Dies führt umgehend zu einer Ansammlung von schneller wachsenden Zellen und damit zu einem Tumor. Weitere Mutationen der DNS und die daraus hervorgehenden Veränderungen in der Zelle selbst ermöglichen die Versorgung des Tumors mit Nährstoffen und das Eindringen von Tumorzellen in umliegende Zellschichten. Damit geht die folgenschwere Bildung von neuen Kolonien im gesamten Körper, sog. Metastasen, einher.
Mutationen, die einen solchen Wachstumsvorteil zur Folge haben, liegen meist in Abschnitten auf der DNS, die zur Herstellung von Proteinen benötigt werden. Eine besondere Rolle für die Tumorentwicklung spielen dabei die Protoonkogene und Tumorsuppressorgene. Proteine, die aus Protoonkogenen entstehen, wirken für die Zellteilung wie das Gaspedal im Auto: sie können die Zellwachstumsgeschwindigkeit beschleunigen. Proteine aus Tumorsuppressorgenen hingegen wirken wie die Bremse und können die Wachstumsgeschwindigkeit reduzieren. So wie die Fahrtgeschwindigkeit des Autos durch Beschleunigen oder Bremsen reguliert wird, so wird die Wachstumsgeschwindigkeit der Zelle durch beide Prozesse entscheidend beeinflusst. Eine Mutation in einem Protoonkogen – dieses wird dadurch zum Onkogen – ist mit einem eingeklemmten Gaspedal vergleichbar, während eine Mutation in einem Tumorsuppressorgen einer defekten Bremse gleichkommt (Abb. 1).

Abb. 1: Die Rolle von Mutationen in Onkogenen und Tumorsuppressorgenen
Mutationen in Onkogenen arbeiten wie das Gaspedal eines Autos, sie erhöhen die Geschwindigkeit der Zellteilung. Mutationen in Tumorsuppressorgenen hingegen wirken wie eine defekte Bremse, bestimmte Haltepunkte werden einfach ignoriert und die Zelle kann sich ohne Kontrolle weiter teilen.
Im normalen, nicht mutierten Zustand bewirken die von diesen Genen kodierten Proteine, dass die Zelle sich zum richtigen Zeitpunkt teilt und in einem Gleichgewicht mit ihrer Umgebung ist. Die Bindung eines Wachstumsfaktors aus dem Blut an Zelloberflächen führt als Auslöser für die Zellteilung zu einer Signalkaskade und schließlich zur Teilung der Zelle. Die Mutation in einem an einer solchen Signalkaskade beteiligten Protoonkogen sorgt für eine dauerhafte Signalgebung zur Zellteilung, auch in Abwesenheit von Wachstumsfaktoren. Oft bildet eine solche dauerhafte Aktivierung aber auch eine Art Achillesferse, da die Tumorzellen von dem onkogenen Signal abhängig werden. Während gesunde Zellen einen Ausfall eines Wachstumssignalweges kompensieren können, sterben die onkogenabhängigen Zellen, sobald das onkogene Signal gehemmt wird.
Signalweiterleitung durch Ras
Eines dieser Protoonkogene der Signalweiterleitung ist Ras. Ras-Mutationen werden in jedem dritten Tumor gefunden. In den besonders aggressiven Krebsarten, wie zum Beispiel dem Bauchspeicheldrüsenkrebs, tragen sogar neun von zehn Tumoren eine Ras-Mutation. Die meisten als Medikament benutzten Moleküle blockieren durch Adsorption an Oberflächenvertiefungen direkt die Aktivität der Zielproteine. Leider besitzt Ras eine sehr glatte Oberfläche, sodass keine Moleküle an ihm haften bleiben. Daher ist es in mehr als 30 Jahren Forschung bis heute nicht geglückt, ein klinisch relevantes Medikament gegen onkogenes Ras zu entwickeln. Für die Signalweiterleitung von Ras ist es bedeutsam, von welchem Aufenthaltsort in der Zelle dieses Signal gegeben wird. Um Signale effizient weiterleiten zu können, muss Ras an Plasmamembranen gebunden sein, da an der Plasmamembran sowohl seine Aktivierung als auch die Signalweiterleitung zur Zellteilung stattfindet. Die Bindung von Ras an die Plasmamembran wird durch nachträglich an das Protein angefügte Modifikationen ermöglicht. Bei Ras ist es der "Fettanker" Farnesyl, ein Fettmolekül, das für die Interaktion mit sämtlichen zellinneren Membranen und der Plasmamembran verantwortlich ist. Onkogenes Ras muss nicht durch externe Signale an der Plasmamembran aktiviert werden, sondern leitet von der Plasmamembran kontinuierlich Signale zur Zellteilung weiter.
Als logische Konsequenz versucht man die Signalweiterleitung durch dauerhaft aktives Ras so zu unterbinden, dass der Prozess der nachträglichen Fettanker-Modifikation und die damit einhergehende Plasmamembran-Lokalisierung blockiert werden. Obwohl die Versuche im Reagenzglas sich als vielversprechend erwiesen hatten, konnte die große Hoffnung, auf diese Weise onkogenes Ras zu inaktivieren, nicht lange aufrechterhalten werden. Die Krebszellen waren in der Lage, die Abwesenheit des Fettankers zu erkennen und dessen Verlust mit einer ähnlichen Modifikation auszugleichen und damit die Plasmamembran-Lokalisierung wieder herzustellen [1].
Räumliche Organisation des Krebsproteins Ras
In verschiedenen Studien erkannte man, dass Ras nach dem Erreichen der Plasmamembran nicht statisch dort verweilt, sondern einem dynamischen Zyklus zwischen Plasmamembran und Zellinnerem unterliegt [2]. Während seiner Reise benötigt es einige Hilfsproteine, die für einen reibungslosen Ablauf des Zyklus sorgen. Eines dieser Hilfsproteine ist der Löslichkeitsfaktor PDEδ. PDEδ sorgt durch die Bindung der Fettanker-Modifikation dafür, dass Ras in einen löslichen Zustand gebracht wird und nicht mit der Vielzahl von zellulären Membranen interagieren kann [3]. Weitere Proteine sorgen für eine Freisetzung des Ras-Proteins von PDEδ in der Nähe einer Ras-Sammelstelle und für den Rücktransport von Ras zur Plasmamembran [4]. In Experimenten mit einer Art molekularbiologischem Baukasten gelingt es, die Menge an Proteinen herunterzuregulieren. Erste Studien zeigten, dass die Absenkung des PDEδ-Proteinlevels zu einer Änderung der Ras-Lokalisation von der Plasmamembran hin zu zellinneren Membranen führte. Diese neue Ras-Verteilung sorgt für ein abgeschwächtes Zellwachstum (Abb. 2).
 zu zellinneren Membranen. PDEδ sorgt dafür, dass es zu einer zentralen Ras-Sammelstelle kommt, von der es wieder zur Zellmembran gelangt. Das links unten eingefügte Bild zeigt eine Mikroskopieaufnahme von leuchtendem Ras in lebenden Zellen mit funktionierendem Ras-Zyklus. Das grün markierte Ras befindet sich an der Zellmembran.Rechte Seite: Hier fehlt der Ras-Interaktionspartner PDEδ. Damit ist weder der Transport zur Ras-Sammelstelle noch der Rücktransport möglich. Ras verweilt somit auf den inneren Zellmembranen. Hier zeigt das rechts unten eingefügte Bild zwei Zellen ohne PDEδ. Das grün markierte Ras befindet sich auf den inneren Membranen der Zelle. Der weiße Balken entspricht einer Länge von 10 µm.")
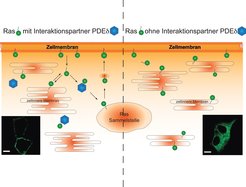
Abb. 2: Räumliche Organisation von Ras
Linke Seite: Ras verlagert über die Zeit seinen Aufenthaltsort von der Zellmembran (Plasmamembran) zu zellinneren Membranen. PDEδ sorgt dafür, dass es zu einer zentralen Ras-Sammelstelle kommt, von der es wieder zur Zellmembran gelangt. Das links unten eingefügte Bild zeigt eine Mikroskopieaufnahme von leuchtendem Ras in lebenden Zellen mit funktionierendem Ras-Zyklus. Das grün markierte Ras befindet sich an der Zellmembran.
Rechte Seite: Hier fehlt der Ras-Interaktionspartner PDEδ. Damit ist weder der Transport zur Ras-Sammelstelle noch der Rücktransport möglich. Ras verweilt somit auf den inneren Zellmembranen. Hier zeigt das rechts unten eingefügte Bild zwei Zellen ohne PDEδ. Das grün markierte Ras befindet sich auf den inneren Membranen der Zelle. Der weiße Balken entspricht einer Länge von 10 µm.
Ein kleines Molekül gegen die Interaktion von Ras mit PDEδ beeinflusst Krebszellwachstum
Die molekularbiologische Herunterregulierung von Proteinen, wie sie im Versuch zuvor genutzt wurde, bietet zurzeit noch keine gezielte Anwendung in der Tumorbehandlung. Um aufzuzeigen, dass PDEδ dennoch ein interessanter Ansatz für die Krebstherapie sein kann, wurde in einem interdisziplinären Forschungsprojekt am Max-Planck-Institut für molekulare Physiologie an einem Inhibitor gearbeitet. Chemiker der Abteilung für Chemische Biologie testeten 150.000 kleine Moleküle im Reagenzglas auf eine Blockade der PDEδ-Ras-Interaktion. Kleine Moleküle, die die Interaktion aufhoben, wurden von Strukturbiologen mittels Röntgenkristallographie im Protein PDEδ sichtbar gemacht und anhand dieser Kristallstrukturdaten in ihrer Affinität modifiziert. Nach einigen Optimierungsprozessen wurde das beste Molekül auf den Namen Deltarasin getauft und seine Wirkung auf Prozesse in der Zelle genauer von der Abteilung für Systemische Zellbiologie untersucht [5].
Mit neuartigen Mikroskopietechniken konnten die Molekularbiologen in lebenden Zellen zeigen, dass die Interaktion zwischen PDEδ und Ras verloren geht, sobald Deltarasin zu den Zellen gegeben wird (Abb. 3a). Im Vergleich zu der vorher erwähnten Methode, bei der die Proteinmenge verändert wird, wird hier der Löslichkeitsfaktor mit dem kleinen Molekül Deltarasin beladen, sodass Ras keinen Platz mehr für die Bindung hat. Der Verlust der PDEδ-Ras-Interaktion führt zum Transport von Ras weg von der Plasmamembran und hin zu den inneren Membranen der Zelle (Abb. 3b). Wachstumsanalysen zeigten, dass die Behandlung mit Deltarasin bei Krebszellen, die eine Ras-Abhängigkeit aufweisen, zum Absterben dieser Zellen führte, während Zellen ohne Ras-Mutation normal weiterwuchsen.
 auf die inneren Membranen der Zelle (nach Deltarasinzugabe).3c. Tumorwachstum im Vergleich zum Anfangsvolumen des Tumors über die Zeit. Die grüne Kurve zeigt die Tumorvolumenzunahme von mit Deltarasin behandelten Mäusen, die schwarze Kurve die von mit Placebo behandelten Mäusen.")
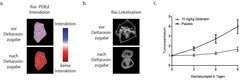
Abb. 3: Einfluss von Deltarasin auf die Ras-PDEδ-Interaktion
3a. Die abgebildete Zelle zeigt vor Deltarasinzugabe eine Interaktion der beiden Proteine. Nach Zugabe von Deltarasin geht diese verloren.
3b. Lokalisationsänderung farbig markierter Ras-Moleküle von der Plasmamembran (vor Deltarasinzugabe) auf die inneren Membranen der Zelle (nach Deltarasinzugabe).
3c. Tumorwachstum im Vergleich zum Anfangsvolumen des Tumors über die Zeit. Die grüne Kurve zeigt die Tumorvolumenzunahme von mit Deltarasin behandelten Mäusen, die schwarze Kurve die von mit Placebo behandelten Mäusen.
Um einen weiteren Schritt in Richtung klinischer Anwendung zu machen, wurde in einer Kollaboration mit Tumorbiologen der Ruhruniversität Bochum die Wirkung von Deltarasin auf von Ras abhängige Bauchspeicheldrüsenkrebszellen im Mausmodell getestet. Hier konnte beobachtet werden, dass die künstlich unter der Haut erzeugten Bauchspeicheldrüsenkrebstumore von mit Deltarasin behandelten Mäusen wesentlich langsamer wuchsen als die Tumore von Mäusen, die mit einem Placebo behandelt wurden (Abb. 3c) [5].
Schlussbemerkung
Durch abteilungsübergreifende interdisziplinäre Zusammenarbeit im Max-Planck-Institut für molekulare Physiologie konnten die Wissenschaftler eine niedrigmolekulare Verbindung finden, das Deltarasin, deren Wirkungsmechanismus einen neuen Ansatzpunkt in der Krebstherapie darstellen kann. Das reduzierte Wachstum der untersuchten Bauchspeicheldrüsenkrebszellen legt den Schluss nahe, dass Deltarasin auch einen Einfluss auf andere Tumorarten mit hoher Ras-Mutationsrate, wie Darmkrebs oder Lungenkrebs, haben könnte.