Forschungsbericht 2013 - Max-Planck-Institut für molekulare Pflanzenphysiologie
Kleine Unterschiede ganz groß – Das Wissen über Sequenzvariabilität erlaubt neue Ansätze in der Pflanzenforschung
Die Sequenzierung pflanzlicher Genome – zentraler Ausgangspunkt der modernen Pflanzenforschung
Als im Jahr 2000 zum ersten Mal die Genomsequenz einer Pflanze veröffentlicht wurde [1], stellte das einen Meilenstein in der Pflanzenforschung dar. Die nunmehr vollständig bekannte genomische Information der Modellpflanze Arabidopsis thaliana (Ackerschmalwand) ermöglichte neue Ansätze zur systematischen Untersuchung der Biologie dieser Spezies und der von Pflanzen im Allgemeinen.
Die Sequenzierung des gesamten Arabidopsisgenoms erforderte in den 1990er-Jahren noch einen erheblichen zeitlichen und finanziellen Aufwand. Die damals verwendete Technologie ging auf Entwicklungen des kürzlich verstorbenen, zweifachen Nobelpreisträgers Frederick Sanger (1918-2013) zurück. 25 Jahre lang war die Sanger-Sequenzierung die dominierende Technologie, bis sie in den vergangenen 10 bis 15 Jahren durch neu entwickelte Sequenziermethoden, zusammengefasst als Next Generation Sequencing Technologies bezeichnet, ersetzt wurde. Diese senkten nicht nur die Kosten der Sequenzierung dramatisch, sondern steigerten zugleich den erzielbaren Durchsatz an Sequenzierungen erheblich. Somit ist es möglich geworden, viele pflanzliche Genome schnell und kostengünstig zu sequenzieren.
Die kleinen Unterschiede – Polymorphismen in pflanzlichen Genomen
Wie menschliche, so unterscheiden sich auch pflanzliche Genome von Individuen ein und derselben Art, solange sie nicht durch vegetative Vermehrung, beispielsweise durch Stecklinge, oder durch wiederholte Selbstbefruchtung, die in einigen Pflanzenarten möglich ist, voneinander abgeleitet sind. Insbesondere weisen Vertreter einer Pflanzenart von verschiedenen geografischen Standorten genetische Unterschiede auf. Denn einige der zufällig entstandenen Genommutationen führten zu günstigen Anpassungen an die jeweiligen Standortbedingungen und setzten sich in den jeweiligen Populationen durch. Von Arabidopsis thaliana sind mehr als Tausend solcher weltweit verbreiteten und standortspezifischen Varianten (Ökotypen) bekannt. Deren jeweilige Genome unterscheiden sich an mehreren Tausend Stellen. Man sagt, sie tragen Sequenzpolymorphismen. Ein Konsortium internationaler Arbeitsgruppen setzte sich in der ersten Dekade des neuen Jahrtausends mit dem sogenannten „1001-Genomprojekt“ zum Ziel, systematisch möglichst viele dieser Ökotypen mit Hilfe der neuen Sequenziermethoden zu entschlüsseln [2]. Über den ersten Ökotyp aus dem Jahr 2000 hinaus [1] sind nunmehr Hunderte weiterer Arabidopsisgenome, die von unterschiedlichen Ökotypen stammen, verfügbar; sie bilden eine ergiebige Grundlage für weitere neue Untersuchungen und Untersuchungsmethoden.
Matapax - Bioinformatik-Plattform für genomweite Assoziationsstudien in Arabidopsis
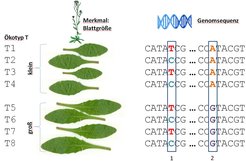
Ein häufig verfolgter Ansatz, die Information über die Sequenzvariabilität gezielt auszunutzen, besteht in sogenannten genomweiten Assoziationsstudien (GWAS). Im Prinzip handelt es sich hierbei um einen Korrelationsansatz, mit dem analysiert wird, welcher Lokus, das heißt, welches Gen oder welcher Genabschnitt und dessen konkrete Sequenz (Allel), mit einem phänotypischen Merkmal korreliert. Man spricht dann von quantitative trait loci (QTLs). Abbildung 1 erläutert schematisch diesen Ansatz.
. Individuelle Ökotypen einer Pflanzenart werden hinsichtlich eines bestimmten Merkmals (zum Beispiel Blattgröße) quantitativ erfasst. Nachfolgend werden Stellen im Genom gesucht, deren Allelausprägung mit diesem Merkmal korreliert. Während in Position 1 keine Ko-Segregation zwischen Blattgröße und Base auftritt, ist sie in Position 2 perfekt. Alle Genome kleinblättriger Pflanzen weisen an Position 2 ein „A“(Adenin) auf, während großblättrige Pflanzen an dieser Position ein „G“ (Guanin) tragen. Somit ist Position 2 ein QTL (quantitative trait locus), und die konkrete Mutation in Position 2 oder weitere Mutationen in der näheren Sequenzumgebung – nahe Bereiche werden gekoppelt vererbt - sind möglicherweise für die unterschiedliche Merkmalsausprägung verantwortlich.")
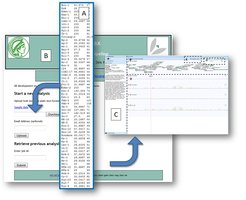
Durch das massive Anwachsen der Sequenzinformation haben GWAS-Ansätze stark an Bedeutung gewonnen. Eine Vielzahl von statistischen GWAS-Computeralgorithmen ist entwickelt worden, die aber oft für Anwender sperrig in der Handhabung sind. Es mangelte an einer webbasierten Ressource, die die Ausführung software-technisch komplexer, aber standardisierbarer Arbeitsschritte - beispielsweise die Formatierung von Eingabedateien, Verwaltung einer Sequenzdatenbank, Installation und Ausführung statistischer Analysesoftware - übernimmt. Mit Matapax hat die Bioinformatikgruppe des Instituts eine solche Online-Ressource geschaffen; sie nimmt dem Anwender aufwendige technische Detailarbeit weitestgehend ab [3]. Anhand der vom Anwender erhobenen quantitativen Merkmalsdaten für bereits sequenzierte Ökotypen führt Matapax alle statistischen Berechnungen aus und präsentiert die Ergebnisse graphisch im Kontext der jeweiligen genomischen Sequenzregionen (Abb. 2). So können schnell Kandidatengene, die vermutlich für die Ausprägung eines Merkmals verantwortlich sind, identifiziert und weiterführende Experimente geplant werden.
 für eine Vielzahl von Ökotypen werden vom Nutzer zu Matapax (B) hochgeladen. Nach erfolgter Analyse werden alle polymorphen Stellen im Genom hinsichtlich ihrer Relevanz für das jeweilige Merkmal bewertet und die Resultate graphisch dargestellt (C).")
Identifizierung neuer regulativer Elemente
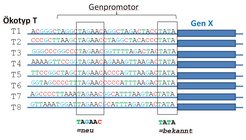
Ein zentraler Ansatz in der bioinformatischen Analyse von Sequenzinformationen ist die gezielte Suche nach konservierten Bereichen. Dabei wird angenommen, dass funktionell wichtige sequenzkodierte Informationen im Laufe der Evolution erhalten bleiben, während Mutationen in anderen Sequenzabschnitten eher toleriert werden. Die Verfügbarkeit einer großen Zahl von Genomen und der darin identifizierten Polymorphismen ermöglicht nun die Anwendung dieses Ansatzes auch auf Ökotypen von Arabidopsis. Spezifisch suchten die Bioinformatiker nach neuen regulativen Elementen, also kurzen Sequenzmotiven, die Genen vorgeschaltet sind und deren Expression beeinflussen [4].
Abbildung 3 illustriert das prinzipielle Vorgehen. Bereits bekannte regulative Elemente (rund 100) wurden tatsächlich als stärker konserviert bestätigt. Im Umkehrschluss sollten somit auch bisher nicht beschriebene Motive, die ebenfalls einen hohen Grad an Sequenzkonservierung aufweisen, ebenso funktionell relevant sein. So konnten 17 neue Kandidatenmotive identifiziert werden, von denen einige vermutlich spezifisch an der Regulation ribosomaler Proteine beteiligt sind. Diese bioinformatisch erhaltenen Ergebnisse müssen nun experimentell überprüft werden.

Enormes Potenzial für weiterführende Forschungen
Die hier erläuterten Beispiele lassen das große Potenzial der nun verfügbaren, umfassenden Genomsequenzinformation erahnen. Die inzwischen angefallene Datenfülle macht die Anwendung computergestützter Analysemethoden zwingend erforderlich. In Kombination mit experimentellen Ansätzen hoffen Pflanzenforscher, mittels computergestützter Genomforschung Erkenntnisse über die Evolution von Pflanzen zu gewinnen und neue Möglichkeiten für die zielgerichtete Züchtung von Nutzpflanzen zu erschließen.