Forschungsbericht 2012 - Max-Planck-Institut für Herz- und Lungenforschung
Quantitative Proteinanalysen mittels Massenspektrometrie
Quantitative Proteomik in lebenden Tieren
Das Proteom beschreibt die Gesamtheit aller in der Zelle vorkommenden Proteine. Im Gegensatz zum statischen Genom ist das Proteom ein dynamisches System, welches zeitlebens durch die Umwelt beeinflusst wird. Neben langfristigen Veränderungen können Zellen aber auch sehr schnell, innerhalb von Sekunden, auf externe Stimuli reagieren. Dies wird vor allem durch Modifikationen an bestimmten Aminosäuren innerhalb der Proteine erreicht. Die bekanntesten posttranslationalen Modifikationen (PTM) sind Phosphorylierungen, Acetylierungen, Glykosylierungen und Ubiquitinierungen. Es hat sich in den letzten Jahren gezeigt, dass die Detektierung und Erforschung solcher Modifikationen einen wichtigen Beitrag zum Verständnis biochemischer Prozesse in lebenden Zellen leistet. Zwar können solche Veränderungen mit verschiedenen immunhistochemischen Methoden nachgewiesen werden, aber eine globale Analyse sämtlicher PTMs ist mit den klassischen Ansätzen nur schwer zu realisieren. In den letzten Jahrzehnten wurde es durch die Entwicklung leistungsfähiger Massenspektrometer möglich, kleine Moleküle, Lipide, Peptide und deren Modifikationen nachzuweisen.
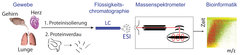
Bei der Proteomforschung kommen vor allem sog. Tandem-Massenspektrometer zum Einsatz, die mit Hilfe von Hochleistungschromatographie (LC: Liquid Chromatography) und Elektrospray-Ionisation (ESI) geladene Peptidmassen messen können (Abb. 1). In einem zweiten Schritt werden diese Peptide im Massenspektrometer durch Gasmoleküle fragmentiert. Dies hat den Vorteil, dass neben der Aminosäuresequenz auch mögliche Modifikationen auf den Peptiden genau lokalisiert werden können. Solche PTMs sind unter anderem bei der Signaltransduktion, der Proteinlokalisierung und der Proteindegradierung extrem wichtig. In den letzten Jahren hat sich gezeigt, dass viele humane Krankheitsbilder gerade auf Veränderungen solcher Modifikationen zurückzuführen sind. So konnte bei einigen metabolischen Syndromen wie zum Beispiel dem Diabetes mellitus II (auch als Alterszucker bekannt) und bei verschiedenen Krebsarten eine Entgleisung von Phosphorylierungsstellen beobachtet werden.
-Flüssigkeitschromatographie getrennt und mittels Elektrospray-Ionisierung (ESI) in das Massenspektrometer gesprüht. Zur Identifizierung der Peptide werden spezielle Proteindatenbanken und Suchalgorithmen verwendet. Abkürzungen: LC liquid chromatography, m/z Masse/Ladung.")
Stabile Isotope für die Proteinquantifizierung in Zellen und Organismen
Eine genaue Proteinquantifizierung ist jedoch mit dem alleinigen Einsatz von Massenspektrometern nur schwer zu realisieren. Als Methode für eine akkurate Mengenbestimmung hat sich die sog. SILAC-Methode (stable isotope labeling of amino acid in cell culture) etabliert. Hier werden Proteine durch den metabolischen Einbau von stabilen, nicht radioaktiven Aminosäure-Isotopen markiert und können somit für die relative Proteinquantifizierung eingesetzt werden. In einem Zellkulturexperiment werden beispielsweise tierische Zellen mit einem markierten Aminosäure-Isotop inkubiert. Nach ca. fünf Zellverdoppelungen sind alle unmarkierten Aminosäuren durch markierte Aminosäuren ersetzt. In den meisten Fällen werden die Aminosäuren Arginin und Lysin verwendet. Nach kompletter Markierung werden die Zellen mit einer unmarkierten Zellpopulation gemischt und für die massenspektrometrische Analyse aufgearbeitet. Veränderte Proteinmengen – Überschüsse an Proteinen – können dann im Massenspektrometer durch die jeweiligen Signalintensitäten der unmarkierten (leicht) und markierten (schwer) Signale bestimmt werden (Abb. 2).
 werden alle Peptide mit einem Lysin um 6 Dalton (Da) schwerer. Nach der Proteinisolierung werden gleiche Mengen der Proteine aus beiden Zellpopulationen (grün und rot) gemischt und im Massenspektrometer analysiert. Das grüne Signal repräsentiert das unmarkierte Peptid, wohingegen das rote Signal das markierte Peptid darstellt. In (a) ist ein schematisches Beispiel für ein typisches SILAC-Paar mit einem 1:1 Verhältnis dargestellt. In (b) ist eine deutliche Abnahme der markierten Signalintensität zu detektieren. Das Verhältnis beider Signale (rot/grün, heavy/light) resultiert in einer relativen Proteinquantifizierung.")
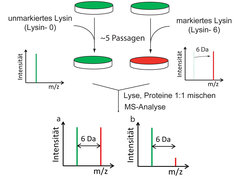
Abb. 2: SILAC-Markierung in der Zellkultur. Die Markierung mit stabilen Isotopen in der Zellkultur wird nach ca. 5 Zellpassagen erreicht. Durch den metabolischen Einbau des markierten Lysins (hier 13C6 Lysin, Lysin-6) werden alle Peptide mit einem Lysin um 6 Dalton (Da) schwerer. Nach der Proteinisolierung werden gleiche Mengen der Proteine aus beiden Zellpopulationen (grün und rot) gemischt und im Massenspektrometer analysiert. Das grüne Signal repräsentiert das unmarkierte Peptid, wohingegen das rote Signal das markierte Peptid darstellt. In (a) ist ein schematisches Beispiel für ein typisches SILAC-Paar mit einem 1:1 Verhältnis dargestellt. In (b) ist eine deutliche Abnahme der markierten Signalintensität zu detektieren. Das Verhältnis beider Signale (rot/grün, heavy/light) resultiert in einer relativen Proteinquantifizierung.
Obwohl diese Technik anfänglich für die Zellkultur entwickelt wurde, konnten Forscher in den letzten Jahren eine Reihe wichtiger Modellorganismen mit solchen Isotopen markieren. Neben einfachen Eukaryoten wie der Bäckerhefe (S. cerevisiae) konnten auch Fliegen (Drosophila), Würmer (C. elegans) und Säugetiere wie die Maus komplett mit SILAC-Isotopen markiert werden [1]. Diese SILAC-Organismen dienen dann als Proteinstandard, um Proteine von unmarkierten Organismen zu quantifizieren (Abb. 3). Ein weiterer wichtiger Modellorganismus für entwicklungsbiologische und medizinische Fragestellungen ist der Zebrafisch (Danio rerio). Aufgrund limitierter Gewebemengen, vor allem bei Fischlarven, ist der Zebrafisch bisher kaum in der Proteomforschung in Erscheinung getreten. Forschern vom Max-Planck-Institut für Herz und Lungenforschung ist es gelungen, eine SILAC-Diät zu entwickeln, die es erlaubt Fische mit Lysin-Isotopen zu markieren. Nach zwei Fischgenerationen konnte ein kompletter Austausch des natürlichen Lysins beobachtet werden. Es ist nun möglich, Proteine und deren posttranslationale Modifikationen durch einfaches Mischen mit dem markierten Standard global zu quantifizieren. Die Entwicklung immer sensitiverer Massenspektrometer und die SILAC-basierte Proteinquantifizierung im Fisch werden es in Zukunft ermöglichen, diesen Organismus auf Proteinebene besser zu verstehen.
Stabile Isotope für die Bestimmung von Proteinhalbwertszeiten

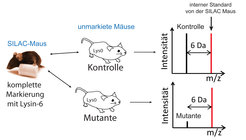
Abb. 3: SILAC in vivo. Die komplett mit Lysin-6 markierte SILAC-Maus fungiert als interner Proteinstandard. Das Mischen des markierten Standards mit den unmarkierten Proteinproben resultiert in einer relativen Quantifizierung zwischen den unmarkierten Tieren. Die SILAC-Maus kann daher als genereller Standard für unmarkierte Mäuse genutzt werden.
Eine weitere wichtige Dimension in der Proteomforschung ist die Kenntnis von Proteinhalbwertszeiten. Lebende Zellen sind offene dynamische Systeme und müssen daher, um ihren Gleichgewichtszustand aufrechtzuerhalten, zeitlebens Proteine neu synthetisieren und entsprechend degradieren. Solche Dynamiken werden seit mehr als 100 Jahren durch sog. Pulse-chase-Experimente mittels radioaktiver Tracer untersucht. In Analogie zu diesen Experimenten können diese Dynamiken heute über die Pulsed-SILAC-Methode gemessen werden. Erste Arbeiten mit stabilen Isotopen in lebenden Organismen wurden schon in den 1930er Jahren von Rudolf Schönheimer durchgeführt [2]. In Kombination mit modernen Massenspektrometern ist es heute für fast alle Proteine möglich, Inkorporationsraten von stabilen Isotopen in neusynthetisierte Proteine zu messen und darüber Proteinhalbwertszeiten zu berechnen. Die Kenntnis solcher Halbwertszeiten ist beispielsweise wichtig, um Proteinstabilitäten bei bestimmten Krankheitsmodellen zu untersuchen (Abb.4).
 verwendet werden.")
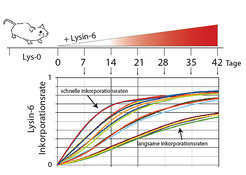
Durch die Gabe von Isotopen-markierten Aminosäuren über die Nahrung werden diese im Körper aufgenommen und in neusynthetisierte Proteine eingebaut. Die Markierungsraten einzelner Proteine können dann mit Massenspektrometern gemessen und zur Berechnung von Proteindynamiken herangezogen werden. Diese Methode wurde unter anderem erfolgreich am grünen Wassermolch (Notophtalmus viridescens) angewendet, der als Modellorganismus für Geweberegeneration dient. Die Arbeitsgruppe von Thomas Braun am MPI für Herz und Lungenforschung konnte durch die Kombination von Herzschädigungen am Molch und SILAC-Markierung eine Reihe neuer Proteine darstellen, die bei der Herzregeneration eine wichtige Rolle spielen [3]. So wurde beispielsweise ein neues Mitglied der sog. CNN-Genfamilie als stark regulierter Faktor bei der Herzregeneration identifiziert. Die extrazellulären CCN-Proteine (Cyr61, CTGF, Nov) spielen eine wichtige Rolle bei der Zellmigration, Matrixproduktion und Zelladhäsion. In Zukunft sollen auch andere regenerierende Modellorganismen wie der Zebrafisch über diesen Pulsed-SILAC-Ansatz auf Proteinebene untersucht werden. Die Wissenschaftler hoffen, so die Mechanismen der Geweberegeneration besser zu verstehen.
Das langfristige Ziel ist es, eine zusammenfassende Darstellung von Proteinmengen, Proteinmodifikationen und Proteinumsatzraten zu etablieren. Weiterhin sollen diese Daten mit mRNA-Expressionsdaten korreliert werden, um die Regulation zwischen Transkriptom und Proteom besser zu verstehen. Dies kann helfen, Krankheitsmodelle genauer zu analysieren und neue diagnostische und therapeutische Strategien für humane Krankheiten zu entwickeln.