Forschungsbericht 2012 - Max-Planck-Institut für Kolloid- und Grenzflächenforschung
Mit Computern den Zuckercode interpretieren
Zucker sorgen für Vielfalt in lebenden Organismen
Jeder Organismus – vom Einzeller bis zum Säuger – ist auf Zucker angewiesen: als Nährstoff und Energielieferant sowie als molekularer Baustein für sehr viele Zellbestandteile. Diese Glycolisierung von beispielsweise Proteinen und Fettsäuren der äußeren Zellmembran übernimmt dabei eine Vielzahl von Aufgaben. Wie Zellen miteinander in Kontakt treten, wie Signalprozesse zwischen und innerhalb von Zellen ablaufen, wie beispielsweise das Immunsystem Eindringlinge erkennt: Alles wird durch die Glycolisierung der beteiligten Biomoleküle mitbeeinflusst oder direkt gesteuert [1].
In welchem Umfang Proteine und Lipide mit Zuckern dekoriert werden, ist nicht direkt im genetischen Code verankert, sie werden bei oder erst nach ihrer Synthese durch spezielle Enzyme angebracht. Die erstaunliche Vielfalt der Zuckerverbindungen, die bisher gefunden wurde, lässt dennoch eine Art Zuckercode vermuten. In der Tat werden viele Zuckerverbindungen spezifisch nach dem Schlüssel-Schloss-Prinzip erkannt und haben definierte Aufgaben. Welche Zucker in welcher Form an Proteine und Antikörper binden, lässt sich heutzutage in modernen Screening-Verfahren mithilfe von sogenannten Glycan-Microarrays im Experiment effizient katalogisieren.
Zucker sind auch Universalwerkzeuge
Das physikalische Erscheinungsbild der meisten Zucker (als Modifikation von Biomolekülen oft auch als Glycane bezeichnet) unterscheidet sich jedoch grundlegend von denen der meisten Proteine. Während diese nach ihrer Synthese und Faltung meist starre, kompakte Formen annehmen, bewahren Glycane einen Großteil ihrer internen Beweglichkeit. Insbesondere durch Fortschritte in Computersimulationen mit atomistischen Modellen ist klar geworden, dass der dynamische Charakter bei der Interpretation von experimentellen Resultaten nicht nur berücksichtigt werden muss, sondern sogar einen Teil der Glycanfunktionalität ausmachen könnte: Man stelle sich etwa einen Schlüssel vor, der gleich zu mehreren Schlössern passt.
So wird auch eine breite Palette eher unspezifischer Funktionen plausibel, die man nicht einfach einer bestimmten chemischen Struktur zuordnen kann. Glycane können die Löslichkeit eines Proteins beeinflussen. Sperrige, langkettige Polysialinlsäuren verhindern bei Embryonen, dass Nervenzellen zu früh aneinanderhaften und vernetzen. Manche Proteine falten sich nicht, wenn man ihre Glycolisierung unterdrückt. Shental-Bechor und Levy [2] haben letzteres Phänomen mit einem stark vereinfachten Computermodell untersucht. Tatsächlich genügt es in diesem Fall, die Glycane lediglich als eine Art Abstandshalter aufzufassen, um ihre Funktion zu verstehen. Die gefaltete Form der betrachteten glycolisierten Proteine wird bevorzugt, weil die ungefaltete destabilisiert ist: Die Zucker sind dann einfach im Weg.
Wozu sind GPI-Anker eigentlich gut?
Will man also ein intuitives Bild eines bestimmten Glycans oder einer Glycolisierung erhalten, das es erlaubt, experimentelle Resultate besser zu verstehen und einzuordnen, ist man auf aussagekräftige numerische und theoretische Modelle angewiesen. Die Anforderungen an Simulation und Theorie sind dabei vielschichtig, und lassen sich gut am Beispiel der sogenannten Glycosylphosphatidylinositol (GPI)-Anker illustrieren.
 Schematischer Aufbau eines GPI-Ankers, eingebettet in eine Membran und mit angehängtem Protein. Das Rückgrat, bestehend aus drei Mannosezuckern (grün) und einem Glucosaminzucker (blauweiß) ist über einen Inositol-Sechsring und eine Phosphatgruppe an den Lipidrest gebunden. (b) GPI-Rückgrat in atomarer Auflösung. Die gelb markierte Atomsequenz charakterisiert die Gesamtkonformation. (c) In einem reduzierten Modell werden die Zuckerringe durch starre Bindungen repräsentiert, drehbare glycosidische Bindungen (rote Segmente) sind die einzigen verbleibenden Freiheitsgrade.")
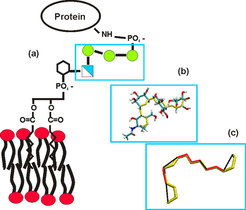
GPI-Moleküle sind Glycane, die ein Protein mit einem Lipidrest verbinden und es so stabil in einer Zellmembran verankern können, siehe Abbildung 1(a). Das relativ kleine Rückgrat ist Bestandteil jedes GPI und kann mit einer Reihe anderer Seitengruppen sowie weiteren, teilweise stark verzweigten Zuckerverbindungen ausgestattet sein. Zwar ist in einigen Fällen bekannt, dass GPI-Moleküle bei Zell-Zell-Kontakt bestimmte Prozesse auslösen, die beispielsweise beim Krankheitsverlauf von Malaria eine wichtige Rolle spielen. Jedoch ist die übergeordnete Bedeutung als Proteinanker bisher noch unverstanden: Es gibt einfachere Möglichkeiten, Proteine an Membrane zu heften [3].
Klassische Molekulardynamiksimulationen reichen nicht aus
Aus theoretischer Sicht beginnt die Untersuchung eines neuen Biomoleküls mit adäquaten atomistischen Modellen, anhand derer die molekulare Dynamik (MD) mittels klassischer Kräfte (elastische-, elektrische-, Adhäsionskräfte) in einer Computersimulation dargestellt werden kann, um etwa das Repertoire der spektroskopischen Strukturanalyse (Röntgenstreuung, Kernspinresonanz) zu unterstützen [4].
 Phage (Virus) an der Oberfläche der äußeren Membran des Bakteriums; grün: Kapsel der Phage mit seiner DNA; gelb: Tail-Spike-Protein (TSP). Die Membran ist mit Lipopolysacchariden durchsetzt, die sich aus Kern ( C ), Lipidrest (Lipid-A) und O-Antigen zusammensetzen. Die Antigene bestehen aus mehreren Wiederholeinheiten (RU) eines Oligosaccharids und bilden in ihrer Gesamtheit einen Schutzmantel. Der Phage erkennt jedoch die Zucker, spaltet sie auf dem TSP und verschafft sich so Zugang zur Membranoberfläche. (b) MD-Simulation der Wechselwirkung von O-Antigenen mit einem TSP des Phagentyps Shigella flexneri 6.")
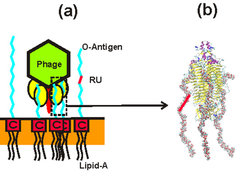
Die erste wirkliche Herausforderung zeigt sich, wenn etwa der GPI-Anker als Ganzes modelliert werden soll: Mit Proteinen, Zuckern und Lipiden befinden sich gleich drei verschiedene Molekülklassen in unmittelbarer Nähe zueinander – klassische Kraftfelder sind aber meist auf einzelne Molekülklassen spezialisiert. Wie lassen sich Parametersätze kombinieren, wo müssen Schnittstellen geschaffen werden? Die nächste Schwierigkeit tritt dann schon bei vermeintlich einfachen Fragen zu den Eigenschaften des Ankers auf. Wie leicht lässt er sich herausziehen, wie beweglich ist das angeheftete Protein, sitzt es direkt auf der Membran oder kann es sich ablösen? Im Gewirr lokaler molekularer Bewegung einer MD-Simulation ist es schwer, das globale Erscheinungsbild eines großen Biomoleküls genau zu charakterisieren. Besonders die erreichbaren Zeitskalen begrenzen die Aussagekraft, wie mit einem anderen Beispiel in Abbildung 2 illustriert wird. Ein Virus durchdringt vor der Infektion eines Bakteriums allmählich dessen schützenden Polysaccharidmantel (a). Mit einer MD-Simulation lässt sich nicht viel mehr als die Adsorption weniger Zuckerketten auf der Proteinoberfläche des Virus erfassen (b). Im Vergleich zum gesamten Prozess eine Momentaufnahme.
Von atomistischer Genauigkeit zur Klarheit
Wie kommt man hier weiter? Mit etwas Einsicht in das Problem kann man angemessen dimensionierte Teile des gesamten Moleküls herausgreifen und separat betrachten. Für das GPI-Rückgrat etwa könnte man fragen, ob es eine starre Verbindung zur Membran herstellt oder eher als flexible Kette aufzufassen ist. Um die innere Steifigkeit des Rückgrats zu charakterisieren, kann man sich dabei auf die leicht drehbaren glycosidischen Bindungen beschränken, siehe Abbildung 1(b), die Zuckerringe selbst sind im Vergleich dazu starr. Auf den reduzierten Satz Variablen lassen sich jetzt alle Umgebungseinflüsse projizieren. Mit anderen Worten: die MD-Methode kann ebenso dazu benutzt werden, um uninteressante Fluktuationen herauszufiltern. Man erhält schließlich ein reduziertes Modell, Abbildung 1(c), das lediglich aus einer vereinfachten Abfolge von Bindungen besteht.
Der vermeintliche Verlust an Information wird durch die Effektivität des reduzierten Modells mehr als wettgemacht. Das Molekül lässt sich nun mit geringem numerischen Aufwand ziehen und drücken, gleichsam spielerisch erkunden, und innerhalb weniger Stunden (anstelle von Wochen) Rechenzeit ist klar, das Rückgrat ist eine eher starre Einheit [5]. Es ist daher unwahrscheinlich, dass ein angeheftetes Protein wie ein Ballon über der Membran schwebt.
Was ist noch zu tun?
Ist die reduzierte Darstellung hinreichend einfach, ist man auch in der Lage, Wechselwirkungen zwischen verschiedenen Komponenten des betreffenden Biomoleküls oder molekularen Komplexes in vereinfachter Form wieder hinzuzufügen. Dabei muss man nicht notwendigerweise wieder auf die Atomistik zurückgreifen. In Levys Modell zum Beispiel werden die Wechselwirkungen der Aminosäuren innerhalb des Proteins schon nicht mehr MD-Studien entnommen, sondern aus der statistischen Verteilung innerhalb des Proteoms abgeleitet.
Im besten Fall werden diese Modelle dann zu einer Art Experimentierkasten, mit dem man bestimmte Situationen effizient durchspielen und Trends entdecken kann. Entscheidend wird hierbei sein, die unterschiedlichen Darstellungsebenen konsistent miteinander zu verbinden. Erst eine entsprechende Leiter aus Experiment, Simulation und Theorie wird es letztlich ermöglichen, die vielseitigen Effekte von Glycanen auf Abläufe innerhalb und außerhalb der Zelle in vollem Umfang zu verstehen. Das Repertoire der theoretischen und numerischen Methoden ist dabei noch lange nicht ausgeschöpft. Wie die Glycobiologie insgesamt ist auch die computergestützte und theoretische Modellierung auf diesem Gebiet eher am Anfang als am Ende ihrer Entwicklung. Dies hat für die Theorie einen besonderen Reiz: Man hat die Gelegenheit, die Welt mit neuen Augen zu entdecken.