Forschungsbericht 2012 - Max-Planck-Institute für experimentelle Medizin
Steuerung des Axonwachstums
Arbeitsgruppe Molekulare und Zelluläre Neurobiologie
Einleitung
Das zentrale Nervensystem (ZNS) gliedert sich in Gehirn und Rückenmark und besteht aus über 100 Milliarden Nervenzellen, den Neuronen, die ein komplexes Netzwerk bilden. Für die Entstehung dieses Netzwerkes ist zunächst die Entwicklung der Neuronenfortsätze, sogenannter Axone, erforderlich. Das Axonwachstum ist präzise gesteuert und während der Entwicklung des ZNS von enormer Bedeutung. Diese Steuerung erfolgt über die Wachstumskegel, die sich am auswachsenden Ende der Axone befinden und wie Sensoren die Umgebung absuchen (Abb. 1). Mit Hilfe von Rezeptoren, die sich am Wachstumskegel befinden, werden inhibitorische (hemmende) und permissive (stimulierende) Signale aus der Umgebung in Axonwachstum und Wegfindung übersetzt. Sowohl inhibitorische als auch permissive Signale sind sog. extrinsische Faktoren, die von außen auf das Axonwachstum des Neurons einwirken.

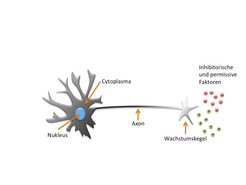
Abb. 1: Der Wachstumskegel
Schematische Darstellung eines Neurons, an dessen sich entwickelndem Axon sich der sensorische Wachstumskegel befindet. Mit diesem Wachstumskegel wird die Umgebung nach permissiven oder inhibitorischen Faktoren abgetastet.Jedoch sind die Mechanismen des Axonwachstums und der Wegfindung nur während der Entwicklung des Zentralen Nervensystems erfolgreich. Kommt es im adulten ZNS zu einer Schädigung der Axone, zum Beispiel einer Verletzung des Rückenmarks, so unterbleibt ein erneutes Auswachsen der Fortsätze und es findet keine Regeneration des Nervengewebes statt. Viele Forschungsstudien sind zu dem Schluss gekommen, dass die Regeneration der Axone aufgrund einer feindlichen Umgebung unmöglich ist. Zum einen verhindert das durch die Verletzung entstandene Narbengewebe das erneute Auswachsen der Axone und zum anderen werden hochgeordnete Strukturen wie das Myelin, das eine Isolierschicht um die Axone bildet, zerstört. Myelinkomponenten, die mit neu auswachsenden Axonen in Kontakt kommen, haben einen starken inhibitorischen Einfluss. Die Wachstumskegel der Axone besitzen den sogenannten Nogo-Rezeptorkomplex, der die Interaktion mit Myelin in ein inhibitorisches Signal übersetzt. Weiterhin stellt die kleine GTPase RhoA einen ganz entscheidenden Effektor in diesem inhibitorischen Signalweg dar. RhoA reguliert das neuronale Cytoskelett, dessen Dynamik für das Axonwachstum notwendig ist.
Zusätzlich zur sich verändernden Umgebung im adulten ZNS ändert sich auch das intrinsische Potenzial der Neurone bezüglich des Axonwachstums. Unter dem intrinsischen Potenzial versteht man Mechanismen, die vom Innern des Neurons heraus auf das Axonwachstum einwirken. Mit zunehmender Reifung sind Neurone nicht mehr in der Lage, das Axonwachstum effizient zu bewerkstelligen. Die dafür verantwortlichen Mechanismen sind allerdings weitgehend unbekannt.
Intrinsische Steuerung des Axonwachstums durch die E3-Ligase Cdh1-APC
Die E3-Ubiquitin-Ligase Cdh1-APC ist ein essenzieller Regulator des Zellzyklus. Während der Zellteilung bindet Cdh1-APC an verschiedene Substratproteine und ubiquitiniert diese, was bedeutet, dass das Substrat mit dem hochkonservierten Protein Ubiquitin verbunden wird. Cdh1 erkennt seine Substrate aufgrund einer sog. D-Box (destruction box), welche die Bindung ermöglicht. Das Proteasom, die Proteinzerkleinerungsanlage der Zelle, erkennt die Ubiquitinierung und leitet den sofortigen Abbau der Substrate ein. Dadurch wird der Übergang zum nächsten Zellzyklusabschnitt sichergestellt.
Cdh1-APC wird allerdings nicht nur in mitotischen Zellen exprimiert, sondern auch in Neuronen, die sich nicht mehr teilen [1]. Mit Hilfe der sog. RNA-Interferenz-Technik (RNAi-Technik) führte dies zu der interessanten Entdeckung, dass Cdh1-APC das Axonwachstum bremst [2]. Die RNAi-Technik ist eine genetische Manipulation der Zelle, bei der die Menge des zu untersuchenden Proteins verringert wird. Eine durch Cdh1-RNAi herbeigeführte reduzierte Menge an Cdh1 stimuliert das Wachstum der Axone von kultivierten Neuronen auf Myelin [2]. Dieses Ergebnis ist deshalb so interessant, weil das Axonwachstum in Neuronen mit normalen Mengen an Cdh1 durch Myelin erheblich blockiert wird. Daraus konnten die Wissenschaftler schließen, dass Cdh1 das Axonwachstum aus dem Inneren des Neurons heraus bremst. Aufgrund der Tatsache dass Cdh1-APC in Neuronen Ligaseaktivität besitzt, die notwendig ist, um das Axonwachstum zu kontrollieren, bestand der nächste Schritt darin, die für den Effekt verantwortlichen Substrate zu finden, um den Cdh1-APC-Signalweg zu etablieren.
Nukleäre Substrate der E3-Ligase Cdh1-APC
Da Cdh1-APC sich hauptsächlich im Nukleus der Neurone befindet und durch seine Aktivität dort das Axonwachstum bestimmt [3], wurde zunächst nach nukleären Substraten gesucht. Cdh1-APC bindet und ubiquitiniert die Transkriptionsfaktoren SnoN und ID2. Sowohl SnoN als auch ID2 stimulieren das Axonwachstum [3, 4]. Beide enthalten eine D-Box, wodurch sie von Cdh1 erkannt und abgebaut werden. Folglich sinkt ihre wachstumsstimulierende Aktivität und das Axonwachstum wird eingeschränkt. Mutiert man die D-Box, wird das Substrat resistent gegenüber Cdh1-APC-vermittelter Ubiquitinierung und Degradierung. Die so angereicherten Substrate SnoN und ID2 entfalten nun ihre Axonwachstums-stimulierende Wirkung.
Cytoplasmatische Substrate der E3-Ligase Cdh1-APC
Obwohl Cdh1-APC vorwiegend im Nukleus präsent und regulativ tätig ist, findet man Cdh1-Aktivität auch im Cytoplasma [3]. Eine besondere Relevanz kommt der gerade erwähnten Beobachtung von Konishi und Kollegen zu, bei der Cdh1-RNAi das Axonwachstum in Gegenwart von Myelin stimuliert [2]. Dies lässt auf eine Kommunikation von Cdh1-APC mit dem inhibitorischen Nogo-Rezeptor-Signalweg und der kleinen GTPase RhoA im Cytoplasma schließen.
Durch ein Schlüsselexperiment wurde die Vermutung unterstützt, dass die kleine GTPase RhoA im Zusammenhang mit Cdh1-APC an der Umsetzung der Inhibierung von Axonwachstum beteiligt ist (Abb. 2A). Weiterhin findet man unter Cdh1-RNAi -Bedingungen eine reduzierte Menge an RhoA am Wachstumskegel der Axone (Abb. 2B). Dies lässt auf eine funktionale Beziehung von Cdh1 und RhoA schließen.
, aktiven (CA) oder inaktiven (DN) Form, so reduziert sich die axonale Länge mit RhoA-WT mäßig und mit RhoA-CA sehr stark.
B. Der Wachstumskegel (weiße Kästchen) in Kontrollneuronen zeigt die Expression der kleinen GTPase RhoA, während diese unter Cdh1-RNAi-Bedingungen nicht mehr nachweisbar ist. GFP (green fluorescent protein) dient der Sichtbarmachung des Neurons.")
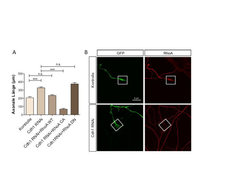
Abb. 2: RhoA ist wichtig für das durch Cdh1-APC inhibierte Axonwachstum
A. Die axonale Länge von Kontrollneuronen wurde verglichen mit Neuronen, in denen Cdh1-RNAi induziert wurde. Dies führt zu längeren Axonen. Exprimiert man nun in Cdh1 RNAi-Neuronen die kleine GTPase RhoA in ihrer wildtypischen (WT), aktiven (CA) oder inaktiven (DN) Form, so reduziert sich die axonale Länge mit RhoA-WT mäßig und mit RhoA-CA sehr stark.
B. Der Wachstumskegel (weiße Kästchen) in Kontrollneuronen zeigt die Expression der kleinen GTPase RhoA, während diese unter Cdh1-RNAi-Bedingungen nicht mehr nachweisbar ist. GFP (green fluorescent protein) dient der Sichtbarmachung des Neurons.Da sowohl RhoA als auch Cdh1 das Axonwachstum inhibieren, war es viel wahrscheinlicher, dass ein RhoA-Regulator, anstelle von RhoA selbst, ein Substrat von Cdh1-APC ist. Um eine Verbindung zwischen Cdh1-APC und RhoA herzustellen, wurde ein Kandidatenscreen durchgeführt, bei dem die E3-Ligase Smurf1 als neuer Bindungspartner von Cdh1 identifiziert wurde [5]. Von Smurf1 war bekannt, dass es RhoA im Cytoplasma ubiquitiniert und seine Proteinmengen reguliert [6]. Weiterhin konnte mit der RNAi-Methode gezeigt werden, dass Smurf1 das Axonwachstum beeinflusst, wofür die Lokalisation von Smurf1 sowohl im Nukleus als auch im Cytoplasma wichtig ist [5]. Smurf1 bindet in einer D-Box-abhängigen Weise an Cdh1 und wird durch Cdh1-APC ubiquitiniert. Die Smurf1-D-Box-Mutante wird wie erwartet stabilisiert und stimuliert das Axonwachstum auch in Gegenwart von Myelin (Abb. 3). Weiterhin konnten die Wissenschaftler zeigen, dass Smurf1-RNAi während der Entwicklung des Cerebellums (Kleinhirn) nicht nur zu kürzeren Axonen und einer unvollständigen Bildung der parallelen Fasern führt, sondern auch einen Migrationsdefekt der Neurone hervorruft. Zusätzlich konnte die Rolle von RhoA in diesem Cdh1-APC/Smurf1-Signalweg etabliert werden. Überexpressions-Experimente ergaben, dass RhoA und vor allem eine stabilisierte Mutante von RhoA die stimulierende Wirkung der Smurf1-D-Box-Mutante auf das Axonwachstum aufheben. Anhand dieser Studie konnte das Repertoire an Cdh1-Substraten erweitert und eine Kommunikation von Cdh1-APC mit extrinsischen Signalwegen demonstriert werden, wobei die kleine GTPase RhoA ein Schlüsselprotein darstellt.
 sind deutlich länger als die Axone auf inhibitorischem Substrat (Myelin). Durch die Expression von Smurf1-DBM3/4 (D-Box-Mutante) wird das Axonwachstum auf Myelin stimuliert und die Länge normalisiert sich. Des Weiteren werden exemplarisch Neurone unter den verschiedenen Bedingungen gezeigt.")
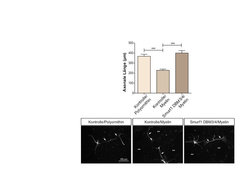
Abb. 3: Smurf1 stimuliert das Axonwachstum
Die Axone der Kontrollneurone auf permissivem Substrat (Polyornithin) sind deutlich länger als die Axone auf inhibitorischem Substrat (Myelin). Durch die Expression von Smurf1-DBM3/4 (D-Box-Mutante) wird das Axonwachstum auf Myelin stimuliert und die Länge normalisiert sich. Des Weiteren werden exemplarisch Neurone unter den verschiedenen Bedingungen gezeigt.Ein weiterer neu identifizierter Bindungspartner von Cdh1 ist das Rho-GTPase-Aktivierungsprotein p250GAP, welches die Inaktivierung von RhoA bewirkt [7]. p250GAP bindet an Cdh1 und stimuliert das Axonwachstum [8]. Im Gegenteil zu Smurf1 scheint p250GAP nicht durch das Proteasom abgebaut zu werden. Ubiquitinierungs-Experimente deuten jedoch auf eine nicht-degradative Ubiquitinierung von p250GAP hin. Unterstützt wird dies durch eine Versuchsreihe, bei der das Fusionsprotein p250GAP-Ubiquitin im Vergleich zum wildtypischen p250GAP nicht in der Lage ist, das Axonwachstum zu stimulieren. p250GAP bindet auch an Smurf1 und reguliert in synergistischer Weise das Axonwachstum. Auch in vivo reguliert p250GAP das Axonwachstum und stimuliert die Migration von Neuronen im Cerebellum. Diese Studie konnte zeigen, dass Cdh1-APC das RhoA-regulierende Protein p250GAP inhibiert und dass RhoA zusammen mit Smurf1 die intrinsische Inhibierung von Cdh1-APC im Cytoplasma umsetzt.
Die Bedeutung der intrinsischen Axonwachstumsregulation

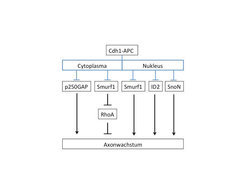
Abb. 4: Schematische Darstellung des Cdh1-APC-Signalwegs
Das Schema zeigt die inhibitorische Wirkung der E3-Ubiquitin-Ligase Cdh1-APC auf verschiedene cytoplasmatische und nukleäre Substrate.Die Arbeiten haben zur Charakterisierung eines neuen intrinsischen Signalwegs der Axonwachstumshemmung geführt, bei dem die E3-Ubiquitin-Ligase Cdh1-APC eine zentrale Rolle spielt. Durch die Inhibierung von mehreren Substraten hält Cdh1-APC das Axonwachstum in Schach (Abb. 4). Die Substrate befinden sich sowohl im Nukleus als auch im Cytoplasma, wo die GTPase RhoA kontrolliert wird. Interessanterweise spielt RhoA also nicht nur eine entscheidende Rolle bei der Wirkung von extrinsischen Inhibitoren, sondern auch bei der intrinsischen Regulation des Axonwachstums. Einerseits offenbart sich mit der Charakterisierung des Cdh1-APC-Signalwegs eine komplexe inhibitorische Regulation des Axonwachstums. Andererseits unterstreicht es die Schwierigkeiten, die überwunden werden müssen, um die axonale Regeneration im zentralen Nervensystem nach einer Verletzung zu stimulieren. Denn zusätzlich zur wachstumsfeindlichen axonalen Umgebung schränken intrinsische Mechanismen das Axonwachstum erheblich ein. In zukünftigen therapeutischen Ansätzen wird es daher notwendig sein, auch die intrinsische Inhibierung zu überbrücken.