Forschungsbericht 2012 - Max-Planck-Institut für Biochemie
Anstandsdamen der Zelle: Rolle in Proteinfaltung und bei der Genese neurodegenerativer Krankheiten
Proteinfaltung und Aggregation
Eiweißmoleküle (Proteine) sind die Träger fast aller zellulären Lebensfunktionen. Sie werden an den Ribosomen als polymere Ketten aus den 20 Aminosäurebausteinen synthetisiert. Um ihre biologischen Funktionen ausüben zu können, müssen die neu synthetisierten Proteinketten eine genau definierte dreidimensionale Konformation einnehmen. Diesen Prozess nennt man Faltung. Er wird wesentlich durch die Abkehr hydrophober Aminosäurebereiche vom wässrigen Milieu, den sogenannten hydrophoben Effekt, angetrieben, wobei letzten Endes aber die Aminosäuresequenz die endgültige dreidimensionale Form des Moleküls bestimmt. Erst in den letzten 20 Jahren wurde erkannt, dass Zellen sogenannte molekulare Chaperone, molekulare Anstandsdamen, enthalten, die den neu synthetisierten Proteinketten bei der Faltung helfen. Diese Chaperone sind selbst Proteine. Ihre Funktionen sind in allen Zelltypen essenziell [1].
Zentrale Aufgabe der Chaperone ist die Verhinderung der Proteinaggregation, also der Verklumpung fehlgefalteter Proteinketten (Abb. 1). Aggregation wird hauptsächlich über hydrophobe Wechselwirkungen ausgelöst. Sie ist besonders bei Proteinen mit komplexer Struktur, zum Beispiel bei Mehrdomänenproteinen, ausgeprägt und wird durch die hohe Proteinkonzentration in der Zelle (300-400 Gramm pro Liter) begünstigt. Aggregierte Proteine stören die Zellfunktion in vielfältiger Weise. Ihre Bildung und Ablagerung ist ursächlich mit neurodegenerativen Krankheiten verbunden.
![Abb. 1: Aggregationsprozesse kompetieren mit produktiver Faltung. Ungefaltete Proteine (U) streben dem nativ gefalteten Zustand (N) zu. Dabei entstehen Faltungsintermediate (I). Die Zustände U und I können Aggregate bilden, besonders im dicht gepackten zellulären Milieu. Ungeordnete Aggregatstrukturen herrschen vor, es entstehen jedoch auch fibrillär geordnete Aggregate (Amyloid) mit zellulärer Toxizität, die ursächlich mit neurodegenerativen Krankheiten verbunden sind. Modifiziert nach [2].](/11588966/original-1508156428.jpg?t=eyJ3aWR0aCI6ODQ4LCJmaWxlX2V4dGVuc2lvbiI6ImpwZyIsIm9ial9pZCI6MTE1ODg5NjZ9--7b134c263838a14812323adc7c68419c938d8a8e "Abb. 1: Aggregationsprozesse kompetieren mit produktiver Faltung. Ungefaltete Proteine (U) streben dem nativ gefalteten Zustand (N) zu. Dabei entstehen Faltungsintermediate (I). Die Zustände U und I können Aggregate bilden, besonders im dicht gepackten zellulären Milieu. Ungeordnete Aggregatstrukturen herrschen vor, es entstehen jedoch auch fibrillär geordnete Aggregate (Amyloid) mit zellulärer Toxizität, die ursächlich mit neurodegenerativen Krankheiten verbunden sind. Modifiziert nach [2].")
![Abb. 1: Aggregationsprozesse kompetieren mit produktiver Faltung. Ungefaltete Proteine (U) streben dem nativ gefalteten Zustand (N) zu. Dabei entstehen Faltungsintermediate (I). Die Zustände U und I können Aggregate bilden, besonders im dicht gepackten zellulären Milieu. Ungeordnete Aggregatstrukturen herrschen vor, es entstehen jedoch auch fibrillär geordnete Aggregate (Amyloid) mit zellulärer Toxizität, die ursächlich mit neurodegenerativen Krankheiten verbunden sind. Modifiziert nach [2].](/11588966/original-1508156428.jpg?t=eyJ3aWR0aCI6MjQ2LCJvYmpfaWQiOjExNTg4OTY2fQ%3D%3D--12749245e6acaed691183fb9f094a96d2afc6c4a)
Abb. 1: Aggregationsprozesse kompetieren mit produktiver Faltung. Ungefaltete Proteine (U) streben dem nativ gefalteten Zustand (N) zu. Dabei entstehen Faltungsintermediate (I). Die Zustände U und I können Aggregate bilden, besonders im dicht gepackten zellulären Milieu. Ungeordnete Aggregatstrukturen herrschen vor, es entstehen jedoch auch fibrillär geordnete Aggregate (Amyloid) mit zellulärer Toxizität, die ursächlich mit neurodegenerativen Krankheiten verbunden sind. Modifiziert nach [2].
Wie funktionieren molekulare Chaperone?
Chaperone verhindern die Aggregation unvollständig gefalteter Proteinketten, indem sie deren exponierte hydrophobe Aminosäuresegmente abschirmen. Da diese Regionen nach erfolgreicher Faltung im Inneren des Proteins verborgen sind, interagieren die Chaperone in der Regel nur so lange, bis der gefaltete, native, Zustand erreicht ist. Die Chaperone können also genau zwischen ungefalteten und gefalteten Proteinen unterscheiden, und dies bei vielen verschiedenen Proteine. Um eine produktive Faltung zu gewährleisten, müssen die Chaperone ihre Klienten allerdings auch wieder loslassen können. Dies geschieht oft in einer ATP-abhängigen Reaktion und mit Hilfe regulierender Kofaktoren. Für die Faltung neu synthetisierter Proteine im Zytosol sind zwei Klassen von Chaperonen von besonderer Bedeutung, die Komponenten der Hsp70 Familie und die zylindrischen Chaperonine [2].
Teamarbeit bei der Proteinfaltung
Prinzipiell unterscheiden wir bei der Faltung neu synthetisierter Proteine zwei Chaperonfunktionen. Kleine Chaperone - zu ihnen zählen Komponenten wie Hsp70 - binden an die naszierenden Ketten, sobald sie aus dem Ribosom austreten. Ihre Aufgabe besteht darin, Ketten voneinander abzuschirmen (Abb. 2). In Zusammenarbeit mit der Forschungsgruppe von Wolfgang Baumeister konnte mit Hilfe der Kryoelektronenmikroskopie gezeigt werden, dass eine besondere Organisationsform der Ribosomen die Wahrscheinlichkeit der Aggregation noch zusätzlich reduziert. Die Ribosomen sind dabei dicht gepackt und so organisiert, dass die Austrittsöffnungen für naszierende Proteinketten den maximal möglichen Abstand zueinander einnehmen [3].
![Abb. 2: Wege der Proteinfaltung im Zytosol bei Bakterien und Eukaryonten. Kleine Chaperone wie der Triggerfaktor (TF) und die Komponenten des Hsp70-Systems (DanK/DnaJ in Bakterien, Hsp70/Hsp40 in Eukaryonten) interagieren mit naszierenden Proteinketten an den Ribosomen und schirmen hydrophobe Segmente ab. In Eukaryonten wird die Funktion von TF möglicherweise durch den Faktor NAC ersetzt. In beiden Systemen werden etwa 10-15% der Proteine zur Faltung an zylindrische Chaperonine (GroEL in Prokaryonten, TRiC in Eukaryonten) weitergeleitet. GroEL kooperiert mit dem Kofaktor GroES, während TRiC von einem solchen Faktor unabhängig ist. Die TRiC Funktion ist mit dem Prozess der Translation durch das Chaperon Prefoldin (PDF) gekoppelt, das sowohl mit naszierenden Proteinketten wie auch mit TRiC interagiert. In Eukaryonten nutzen Proteine der Signaltransduktion (Kinasen) das Chaperon Hsp90 zur Faltung und konformationellen Regulation. N, nativ gefaltetes Protein. Modifiziert nach [2].](/11588950/original-1508156428.jpg?t=eyJ3aWR0aCI6ODQ4LCJmaWxlX2V4dGVuc2lvbiI6ImpwZyIsIm9ial9pZCI6MTE1ODg5NTB9--100d7d0ec2c8568dad8ee87041a3f79813a1c26b "Abb. 2: Wege der Proteinfaltung im Zytosol bei Bakterien und Eukaryonten. Kleine Chaperone wie der Triggerfaktor (TF) und die Komponenten des Hsp70-Systems (DanK/DnaJ in Bakterien, Hsp70/Hsp40 in Eukaryonten) interagieren mit naszierenden Proteinketten an den Ribosomen und schirmen hydrophobe Segmente ab. In Eukaryonten wird die Funktion von TF möglicherweise durch den Faktor NAC ersetzt. In beiden Systemen werden etwa 10-15% der Proteine zur Faltung an zylindrische Chaperonine (GroEL in Prokaryonten, TRiC in Eukaryonten) weitergeleitet. GroEL kooperiert mit dem Kofaktor GroES, während TRiC von einem solchen Faktor unabhängig ist. Die TRiC Funktion ist mit dem Prozess der Translation durch das Chaperon Prefoldin (PDF) gekoppelt, das sowohl mit naszierenden Proteinketten wie auch mit TRiC interagiert. In Eukaryonten nutzen Proteine der Signaltransduktion (Kinasen) das Chaperon Hsp90 zur Faltung und konformationellen Regulation. N, nativ gefaltetes Protein. Modifiziert nach [2].")
![Abb. 2: Wege der Proteinfaltung im Zytosol bei Bakterien und Eukaryonten. Kleine Chaperone wie der Triggerfaktor (TF) und die Komponenten des Hsp70-Systems (DanK/DnaJ in Bakterien, Hsp70/Hsp40 in Eukaryonten) interagieren mit naszierenden Proteinketten an den Ribosomen und schirmen hydrophobe Segmente ab. In Eukaryonten wird die Funktion von TF möglicherweise durch den Faktor NAC ersetzt. In beiden Systemen werden etwa 10-15% der Proteine zur Faltung an zylindrische Chaperonine (GroEL in Prokaryonten, TRiC in Eukaryonten) weitergeleitet. GroEL kooperiert mit dem Kofaktor GroES, während TRiC von einem solchen Faktor unabhängig ist. Die TRiC Funktion ist mit dem Prozess der Translation durch das Chaperon Prefoldin (PDF) gekoppelt, das sowohl mit naszierenden Proteinketten wie auch mit TRiC interagiert. In Eukaryonten nutzen Proteine der Signaltransduktion (Kinasen) das Chaperon Hsp90 zur Faltung und konformationellen Regulation. N, nativ gefaltetes Protein. Modifiziert nach [2].](/11588950/original-1508156428.jpg?t=eyJ3aWR0aCI6MjQ2LCJvYmpfaWQiOjExNTg4OTUwfQ%3D%3D--0b4ed151174a75f487466af4f01c376b5c22b6f6)
Hsp70-Chaperone bestehen aus zwei funktionelle Domänen, eine N-terminale ATPase-Domäne und eine C-terminale Peptidbindungs-Domäne. Der Zugang in die Peptidbindetasche wird durch ein verstellbares a-helikales Segment reguliert, dessen Position von der ATPase-Domäne gesteuert wird. Im ATP gebundenen Zustand ist die Tasche offen, während sie im ADP-Zustand geschlossen ist. Die ATP Hydrolyse an Hsp70 wird durch den Kofaktor Hsp40 stark beschleunigt, was zum Schließen der Peptidbindetasche führt. Ein Nukleotid Austauschfaktor (NEF; GrpE in Bakterien) löst dann die Dissoziation des gebundenen ADP aus, woraufhin erneute ATP Bindung die Freisetzung der Proteinkette induziert und den Reaktionszyklus abschließt. In jedem Bindezyklus wird dem Proteinsubstrat die Gelegenheit zur Faltung, das heißt zum Verbergen hydrophober Bereiche gegeben. Ist dies erfolgreich, so erfolgt keine Rückbindung an Hsp70. Im anderen Fall durchläuft die naszierende Kette einen weiteren Bindezyklus.
Das Hsp70 Chaperon nimmt in fast allen Zelltypen eine zentrale Stellung bei der Proteinfaltung ein. Besonders stark aggregationsgefährdete Proteine sind jedoch auf die Hilfe der Chaperonine angewiesen.
Chaperonine - Nanokäfige für die Proteinfaltung
Die Chaperonine (Hsp60) gehören zu den faszinierendsten ATP getriebenen molekularen Maschinen. Sie bilden Hohlkomplexe, die ein einzelnes, noch ungefaltetes Protein in ihrer zentralen Kammer einschließen und ihm somit die Faltung unter Bedingungen erlauben, bei denen Aggregation ausgeschlossen ist (Abb. 3). Dieser Mechanismus ist am besten für das bakterielle Chaperonin GroEL und seinen Kofaktor GroES verstanden [1].

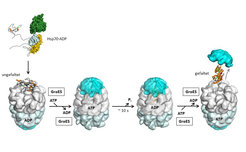
GroEL besteht aus zwei heptameren Ringen aus 60 kDa Untereinheiten, die Rücken an Rücken aufeinandergestapelt sind. Die Ringöffnung exponiert hydrophobe Aminosäurereste für die Bindung ungefalteter Proteine. GroES ist ein heptamerer Ring aus 10 kDa Untereinheiten, der sich wie ein Deckel auf die Ringöffnung des GroEL legt. GroES bindet bevorzugt an nur einen der Ringe, und zwar an denjenigen, der das Proteinsubstrat enthält. Dieser Schritt führt zum Einschluss des gebundenen Proteins in der zentralen Kavität des GroEL. Diese nimmt sodann die Funktion eines Faltungskäfigs an, der Proteine bis zu einer Größe von 60 kDa aufnehmen kann [1]. Das Substratprotein bleibt für etwa 10 Sekunden eingeschlossen, die Zeit, die für die Hydrolyse der 7 ATP Moleküle im GroEL-Ring benötigt wird. Erst danach kann die ATP-Bindung am gegenüberliegenden Ring die GroES-Dissoziation und Ringöffnung auslösen. Ein bereits gefaltetes Protein wird freigesetzt, während unvollständig gefaltete Moleküle rückgebunden werden und erneut einen Faltungszyklus durchlaufen.
Es ist leicht zu verstehen, wie dieser Mechanismus die Proteinfaltung unter Ausschluss der Aggregation gewährleistet. GroEL-GroES stellt sozusagen ein Mini-Reagenzglas für ein einzelnes Molekül dar. Unsere neueren Untersuchungen haben jedoch gezeigt, dass dieses Modell unvollständig ist, denn das Reagenzglas ist eher ein Nano-Käfig, in den das sich faltende Protein sozusagen eingezwängt wird. Dies hat eine Änderung der Energielandschaft der Faltungsreaktion zur Folge, wobei die räumliche Einengung zur bevorzugten Bildung kompakter Formen führt. Tatsächlich kann der Einschluss in den Faltungskäfig zu einer erheblichen Beschleunigung der Faltung im Vergleich zur Spontanfaltung in freier Lösung führen [4, 5].
Rolle der Chaperone bei neurodegenerativen Faltungskrankheiten
Trotz aufwändiger Proteinqualitätskontrolle entstehen in unseren Zellen ständig fehlerhafte Proteine. Die Fehlfaltung und Aggregation von Proteinen wird zunehmend als die Ursache wichtiger Erkrankungen erkannt. Hierzu gehören eine Reihe altersabhängiger neurodegenerativer Syndrome wie Morbus Alzheimer, Parkinson und Chorea Huntington. Ihr zentrales zellpathologisches Merkmal ist die Bildung und Ablagerung von fibrillären Proteinaggregaten, sogenanntem Amyloid, im Gehirn, entweder innerhalb oder außerhalb der Zellen (Abb. 4). Ihre Bildung ist untrennbar mit Toxizitätsphänomenen verbunden, deren exakte Mechanismen noch nicht verstanden sind.
![Abb. 4: Verhinderung der pathologischen Proteinaggregation durch pharmakologische Chaperonaktivierung. Säugerzellen (links), die das Krankheitsprotein von Chorea Huntington in fluoreszenzmarkierter Form synthetisieren, enthalten große Aggregate (grün) neben dem Zellkern (blau). Behandlung der Zellen mit steigenden Konzentrationen von Geldanamycin (GA; 18 nM bzw. 180 nM) führt zur Aktivierung der Chaperone und Verhinderung der Aggregation. Das fluoreszierende Protein ist nun gleichmäßig über die gesamte Zelle verteilt. Modifiziert nach [9].](/11590726/original-1508156499.jpg?t=eyJ3aWR0aCI6ODQ4LCJmaWxlX2V4dGVuc2lvbiI6ImpwZyIsIm9ial9pZCI6MTE1OTA3MjZ9--7184bc76517a84bb03535bdf87a3dceaaf4def12 "Abb. 4: Verhinderung der pathologischen Proteinaggregation durch pharmakologische Chaperonaktivierung. Säugerzellen (links), die das Krankheitsprotein von Chorea Huntington in fluoreszenzmarkierter Form synthetisieren, enthalten große Aggregate (grün) neben dem Zellkern (blau). Behandlung der Zellen mit steigenden Konzentrationen von Geldanamycin (GA; 18 nM bzw. 180 nM) führt zur Aktivierung der Chaperone und Verhinderung der Aggregation. Das fluoreszierende Protein ist nun gleichmäßig über die gesamte Zelle verteilt. Modifiziert nach [9].")
![Abb. 4: Verhinderung der pathologischen Proteinaggregation durch pharmakologische Chaperonaktivierung. Säugerzellen (links), die das Krankheitsprotein von Chorea Huntington in fluoreszenzmarkierter Form synthetisieren, enthalten große Aggregate (grün) neben dem Zellkern (blau). Behandlung der Zellen mit steigenden Konzentrationen von Geldanamycin (GA; 18 nM bzw. 180 nM) führt zur Aktivierung der Chaperone und Verhinderung der Aggregation. Das fluoreszierende Protein ist nun gleichmäßig über die gesamte Zelle verteilt. Modifiziert nach [9].](/11590726/original-1508156499.jpg?t=eyJ3aWR0aCI6MjQ2LCJvYmpfaWQiOjExNTkwNzI2fQ%3D%3D--56997d203f5f22ced0dee90fea09de3e89f32b0a)
Wir haben uns zunächst der Frage zugewandt, warum es trotz des Vorhandenseins der Chaperone zu toxischen Proteinaggregaten kommen kann. Interessanterweise konnten wir zeigen, dass insbesondere Chaperone der Hsp70 Familie durchaus in der Lage sind, die pathologische Proteinaggregation zu verhindern [6, 7]. Sie agieren dabei in der Frühphase der Aggregation, indem sie die sogenannte Nukleation der Aggregate verhindern [6]. Dies wird aber nur beobachtet, wenn die Chaperone aktiviert und in ausreichender zellulärer Konzentration vorliegen (vgl. Abb. 4). Das neurodegenerative Aggregationsphänomen scheint sich also als Konsequenz einer unzureichenden Chaperonkapazität zu manifestieren. Tatsächlich deuten neuere Befunde darauf hin, dass es im Rahmen des Alterungsprozesses zu einer Abnahme der Funktionalität der Chaperone kommt [1]. Dies dürfte erklären, warum die genannten neurodegenerativen Krankheiten altersabhängig auftreten.
Eine weitere zentrale Frage betrifft die Mechanismen, durch die die Aggregate Zelltoxizität auslösen. Wir haben uns diesem Problem unter Verwendung eines systematischen Ansatzes gewidmet. Dabei wurde insbesondere die Hypothese getestet, dass die toxischen Aggregate – oder ihre oligomeren Vorstufen – andere zelluläre Proteine koaggregieren und auf diese Weise deren Funktion stören. Wir konnten mit Hilfe quantitativer Proteomikmessungen zeigen, dass die Aggregate, neben verschiedenen Chaperonen, mit zahlreichen (ca. 100) endogenen Proteinen interagierten [8]. Diese Proteine sind zumeist verschiedenen Schlüsselfunktionen der Zellregulation zuzuordnen. Die Toxizität der Aggregate beruht also, zumindest zum Teil, auf ihrer Fähigkeit, multiple Proteine zu sequestrieren und somit funktionell zu inhibieren.
Ausblick
Bei der weiteren Erforschung der Proteinfaltung wird es darauf ankommen, Wissen und Analysemethoden aus verschiedenen Disziplinen zusammenzuführen. Zunehmend werden wir in der Lage sein, raffinierte biophysikalische Techniken zur zeitaufgelösten Analyse der Proteinkonformation auf zelluläre Verhältnisse anzuwenden mit dem Fernziel, Einzelmoleküle während der Synthese und Faltung in der Zelle verfolgen zu können. Eine große Herausforderung im medizinischen Bereich besteht darin, die Struktureigenschaften von Proteinaggregaten zu entschlüsseln, die ihnen toxische Eigenschaften verleihen. Die Aufklärung der molekularen Mechanismen neurodegenerativer Krankheiten ist besonders in den Industriestaaten mit alternder Bevölkerungsstruktur von großer Dringlichkeit. Anlass zu Optimismus geben Befunde, nach denen das zelleigene Chaperonsystem zur Aggregationsverhinderung pharmakologisch aktiviert werden kann ([9]; Abb. 4).