Forschungsbericht 2009 - Max-Planck-Institut für molekulare Physiologie
Entwicklung chemischer Sonden zur Proteasenforschung
Einleitung
Proteasen katalysieren die Hydrolyse von Amidbindungen in Proteinen. Neben dem Verdau von Proteinen zur Bereitstellung von Aminosäurebausteinen für den Aufbau neuer Proteine dient die Proteolyse vor allem der Regulation biologischer Prozesse. Bekannte Beispiele hierfür sind die so genannte limitierte Proteolyse, welche inaktive Vorläuferproteine in die reifen aktiven Proteine überführt oder aber die regulierte Proteolyse, in welcher spezifische Proteine durch einen Verdau irreversibel deaktiviert werden.
Interessanterweise sind in nahezu jedem biologischen Prozess spezifische, proteolytische Schritte vorhanden. Um diese mit der notwendigen Selektivität auszustatten, werden eine Vielzahl unterschiedlicher Proteasen in jeder Zelle benötigt. So codiert das menschliche Genom für ca. 500 Proteasen, das entspricht in etwa 2 % aller codierten Proteine. Die proteolytische Aktivität jeder Protease unterliegt einer strikten Regulation; eine gelegentlich auftretende Fehlregulation von Proteasen führt häufig zu schwerwiegenden Erkrankungen wie z. B. Krebs oder Alzheimer. Ein tiefergehender Einblick in die Regulation und Funktion von Proteasen ist daher von großem Interesse für die biologische und medizinische Forschung.
Um Proteasen in ihren komplexen zellulären Kontexten studieren zu können, bedarf es der Entwicklung geeigneter neuer Hilfsmittel. Dazu gehören die hier vorgestellten chemisch-biologischen Methoden. Ziel dieser Arbeiten ist die Synthese selektiver chemischer Sonden, mit denen gezielt ausgewählte Proteasen innerhalb des komplexen Proteoms studiert werden können. Dabei verfolgen die Wissenschaftler in Dortmund zwei unterschiedliche Ansätze: Einerseits werden chemische Hilfsmittel für biologisch interessante Proteasefamilien unter Zuhilfenahme von z.B. strukturbasiertem Design entwickelt. Andererseits dienen biologisch-aktive Substanzen wie beispielsweise bakterielle Naturstoffe, deren genaue biologische Funktion jedoch vor Beginn dieser Arbeiten noch nicht aufgeklärt war, als Leitstrukturen zur Entwicklung strukturell und funktionell neuartiger Sonden. Ebenso wird untersucht, ob die synthetisierten Verbindungen auch für medizinische Anwendungen geeignet sind. Für diese Ansätze sollen hier kurz zwei Fallbeispiele dargestellt werden.
Syringoline und Syrbactine
Einige Stämme des Pflanzenpathogens Pseudomonas syringae pv. syringae sekretieren unter Infektionsbedingungen eine Klasse von Naturstoffen, die unter dem Namen Syringoline bekannt sind (Abb. 1a). Die Biosynthese dieser strukturell interessanten, peptidischen Naturstoffe verläuft über eine gemischte nicht-ribosomale Peptid- und Polyketidsynthese. Ihre biologische Rolle, ihr Wirkmechanismus und ihr Potenzial als mögliche Wirkstoffe für chemisch-biologische und medizinische Untersuchungen waren jedoch zu Beginn dieser Arbeiten noch nicht bekannt.
 Chemische Strukturen der Syringolin-Peptidwirkstoffe Die absolute Stereochemie der chiralen Zentren konnte erst durch diese Arbeiten zugeordnet werden. b) Chemische Strukturen der bisher bekannten Glidobactine Auf der linken Seite ist die chemische Struktur von Glidobactin A zu sehen (GlbA), auf der rechten Seite die weiteren, bisher bekannten Glidobactine bzw. Cepafungine. Zusammen mit den Syringolinen bilden diese die neue Naturstoffklasse der Syrbactine.")
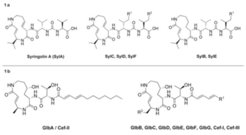
Es konnte nun gezeigt werden, dass der Hauptmetabolit der Syringoline, Syringolin A, während der Infektion von Pflanzen als bakterieller Virulenzfaktor dient [1]. Die beobachtete Virulenz wird durch eine Hemmung des 20S-Pflanzenproteasoms erreicht. Das 20S-Proteasom ist eine multikatalytische, hochkomplexe Protease, welche eine Schlüsselrolle in der regulatorischen Proteolyse in eukaryotischen Zellen spielt. Dies bedeutet, dass die Syringolin A-produzierenden Pathogene als Infektionsstrategie die Abschaltung eines zentralen Regulationsmechanismus der Pflanze verwenden.
Des Weiteren konnten die Forscher zeigen, dass nicht nur Syringoline, sondern auch die strukturell verwandten Glidobactine (Abb. 1b) eine 20S-Proteasomhemmung mittels eines analogen molekularen Mechanismus herbeiführen. Aus diesen Gründen wurden Syringoline und Glidobactine zu einer neuen Strukturklasse von Proteasom-Inhibitoren, die den Namen Syrbactine erhielt, zusammengefasst.
Mechanismus der Proteasomhemmung
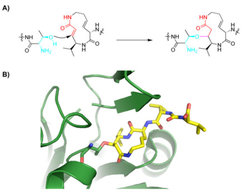
Von entscheidender Bedeutung für die Hemmung ist eine α,β-ungesättigte Amidgruppe im Makrozyklus des Syringolins. Diese Gruppe wird von der katalytisch aktiven Hydroxylgruppe des Threonins modifiziert, das im aktiven Zentrum des Proteasoms lokalisiert ist. Das hieraus resultierende kovalente Konjugat ist unter physiologischen Bedingungen stabil und bewirkt somit eine irreversible Ausschaltung des gesamten proteolytischen Systems (Abb. 2).
 Nucleophiler Angriff auf das α,β-ungesättigte Amidsystem von Syringolin A führt zu einer irreversiblen Hemmung. b) Eine Röntgenstrukturanalyse des 20S-Hefeproteasoms im Komplex mit dem Naturstoff Syringolin A belegt den postulierten, molekularen Wirkmechanismus.")
Trotz ihrer strukturellen Ähnlichkeit zeigen Syringolin A und Glidobactin A ein unterschiedliches Hemmprofil bezüglich der drei unterschiedlichen proteolytischen Zentren des 20S-Proteasoms. Während Syringolin A alle drei proteolytischen Aktivitäten inhibiert, hemmt Glidobactin A nur die chymotryptische und tryptische Aktivität, diese jedoch mit insgesamt höherer Affinität. Eine Strukturanalyse verschiedener Syrbactine deutete an, dass die unterschiedliche Affinität auf die trotz aller strukturellen Ähnlichkeiten leicht unterschiedliche Geometrie des Makrozyklus zurückzuführen sein könnte, während die höhere Aktivität von Glidobactin A durch die Adressierung einer konservierten lipophilen Tasche mittels einer stark lipophilen Seitenkette erklärt werden konnte. Die gezielte Synthese und der Einsatz eines Syringolin-A-Derivates zeigte dann, dass diese Strukturmerkmale in der Tat die molekulare Basis der beobachteten unterschiedlichen Wirkaktivitäten darstellen [2].
Zur Darstellung von Syringolinen existieren mehrere Synthesewege. Es zeigte sich, dass zur Synthese der stark Ring-gespannten Syringolin-A-Derivate eine Ringschlussmetathese (RCM) als Schlüsselschritt besonders geeignet ist. Mittels dieser etablierten Syntheseroute wurden in der Zwischenzeit mehrere Syringolin-A-Derivate für weitere biologische, aber auch medizinische Untersuchungen hergestellt [2].
Selektivität der Proteasomhemmung

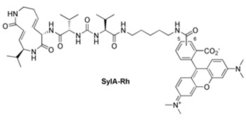
Eine wichtige Frage ist die Selektivität der Syringolin-20S-Proteasomhemmung in einem komplexen Proteom. Um diese zu beantworten, wurde ein fluoreszierendes Syringolin-A-Derivat synthetisiert und mit diesem Markierungsstudien ausgewählter Proteome durchgeführt (Abb. 3). [3] Diese Studien zeigten eine beeindruckende Selektivität der Proteasomhemmung, da nur Untereinheiten des 20S-Proteasoms durch diese Sonde markiert wurden.
Medizinische Anwendung der Syringoline
Es wurden erste Studien zur Evaluierung einer möglichen medizinischen Anwendung der Syringoline durchgeführt. Die Hemmung des Proteasoms ist eine etablierte Strategie im Kampf gegen bestimmte Krebserkrankungen wie z. B. der multiplen Myelose. Zur Chemotherapie dieser Krebserkrankung ist seit einiger Zeit bereits ein erster Proteasom-Inhibitor mit dem Namen Bortezomib zugelassen, dessen Anwendung jedoch starke Nebenwirkungen verursacht und darüber hinaus nach einiger Zeit Resistenzen hervorruft. In einer ersten Studie mit dem fluoreszierenden Syringolin-A-Derivat zeigte sich, dass Syringolin A auch das Proteasom in Bortezomib-adaptierten Zelllinien hemmt und somit eine interessante Leitstruktur für die Entwicklung eines neuen Chemotherapeutikums darstellt [3]. Eine mögliche Anwendung von Syrbactinen als Antikrebsmittel konnte in einer zweiten Studie bestätigt werden, in welcher der Effekt ausgewählter Syrbactine auf Krebszelllinien untersucht wurde.
Zurzeit wird in weitergehenden Studien untersucht, welche Rolle das gezielte Abschalten des Ubiquitin-Proteasom-Systems durch Syringoline im Zusammenhang mit der Besiedlung der Pflanze durch Pathogene spielt. Darüber hinaus werden weitere Derivate von Syringolin A hergestellt, um deren klinische Relevanz evaluieren zu können.
Die biologische Rolle der HtrA-Proteasen: Aufklärung durch chemische Tools
HtrA-Proteasen stellen eine einzigartige Familie weitgehend konservierter Serinproteasen dar, die aus einer Chymotrypsin-artigen Protease- und einer oder auch zwei PDZ-Domänen aufgebaut sind. Eine PDZ-Domäne ist ein Teil eines Proteins, der mit anderen Proteinen interagieren kann. Vertreter dieser Serinproteasen finden sich in allen bekannten Spezies. Die meisten Untersuchungen dieser Proteasefamilie wurden bisher mit bakteriellen Organismen, insbesondere Escherichia coli, durchgeführt. In Bakterien sind die zwei HtrA-Proteasen DegS und DegP an der periplasmatischen Proteinqualitätskontrolle beteiligt. DegS übernimmt dabei die Rolle eines Stress-Sensors, der z. B. aktiviert wird, wenn fehllokalisierte Proteine im Periplasma auftreten, wodurch eine Stressantwort des Systems ausgelöst wird. DegP hingegegen übernimmt den Abbau fehlgefalteter oder auch fehllokalisierter Proteine im Periplasma.
Das große Interesse an HtrA-Proteasen gründet sich auf den ungewöhnlichen Aktivierungsmechanismus dieser Proteasen. Im Gegensatz zu herkömmlichen Serinproteasen, welche nach Aktivierung durch proteolytische Prozessierung intrinsisch aktiv bleiben, können HtrA-Proteasen reversibel zwischen aktiven und inaktiven Zuständen umschalten. [4]
Zu Beginn dieser Arbeiten war dies jedoch nur für die HtrA-Protease DegS gezeigt worden. Mittels Röntgenstrukturanalysen in Verbindung mit biochemischen Assays konnte für diese Protease der Aktivierungsmechanismus aufgeklärt werden: Durch C-terminale Bindung peptidischer Liganden an die PDZ-Domäne nimmt die vorletzte Aminosäure des Aktivierungsliganden eine solche Position ein, dass diese in eine konservierte Tasche der Proteasedomäne bindet, wodurch eine Umordnung der Proteasedomäne in eine proteolytisch aktive Proteasedomäne bewirkt wird. Aufgrund der räumlichen Trennung der Umorganisation des aktiven Zentrums von der Bindungsstelle des aktivierenden Ligandens handelt es sich somit um eine allosterische Aktivierung.


Ausgehend von diesen Studien wurde untersucht, ob die allosterische Aktivierung durch Bindung eines geeigneten Liganden an die PDZ-Domäne ein allgemeiner, konservierter Mechanismus innerhalb der HtrA-Proteasenfamilie ist oder spezifisch nur bei DegS auftritt. Als weitere HtrA-Protease haben wir hierzu die Escherichia coli -HtrA-Protease DegP untersucht.
Zuerst musste ein geeigneter Assay zur Bestimmung der proteolytischen Aktivität von DegP etabliert werden. In einem ersten Schritt wurden geeignete Substrate und Inhibitoren für DegP mittels rationalem Design entworfen. [5] Biochemische Assays von DegP und DegP-Mutanten zeigten anschließend, dass die allosterische Aktivierung von DegP analog zu DegS verläuft (Abb. 4). So binden allosterische Regulatoren an die PDZ1-Domäne von DegP und bewirken hierdurch eine Aktivierung der Protease. [6]
Die allosterische Aktivierung als neue Komponente im Stress-Signalweg
, wodurch eine proteolytische Kaskade bestehend aus den Proteasen DegS, RseP und ClpXP initiiert wird und in der Freisetzung des Faktors RpoE endet. Dieser bewirkt, neben vielen weiteren Effekten, eine Expression der HtrA-Protease DegP. Neben dieser über DegS verlaufenden, transkriptorischen Stressantwort zeigen unsere Arbeiten, dass auch noch ein direkter, allosterischer Regulationsmechanismus, der eine Aktivierung der HtrA-Protease DegP bewirkt, existiert (gewölbte Pfeile).")
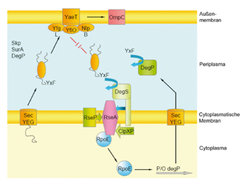
Diese allosterische Aktivierung trägt vermutlich zur Regulation der Stressantwort im Periplasma von Bakterien bei. Neben der bekannten, über DegS initiierten Stressantwort erlaubt die allosterische Regulation eine schnelle, energieeffiziente und robuste Antwort im Periplasma. Der entsprechende Stress-Signalweg sollte um diese Regulationskomponente erweitert werden (Abb. 5).
Umorganisation der HtrA-Proteasedomäne durch kleine Moleküle
Zur chemisch-biologischen Untersuchung der HtrA-Proteasen wären nicht-peptidische, „klassische“ kleine Moleküle von Vorteil, welche z. B. durch systematisches Screening von Substanzbibliotheken aufgefunden werden könnten. Voraussetzung hierfür ist jedoch, dass bereits kleine Moleküle die notwendige allosterische Umorganisation der Proteasedomäne der HtrA-Proteasen induzieren. Um dies zu zeigen, wurde eine Modellstudie an der strukturell einfacheren HtrA-Protease DegS durchgeführt. Bereits chemische Substanzen mit einem Molekulargewicht MG
Zurzeit werden Hochdurchsatz-Screenings zur Identifizierung allosterischer Aktivatoren und auch Inhibitoren einzelner HtrA-Proteasen durchgeführt. So gelang es bereits, einen vielversprechenden niedermolekularen Naturstoff aufzufinden, welcher als reversibler Aktivator der menschlichen HtrA-Protease HtrA1 wirkt. Dieser wird jetzt in weitergehenden Studien auf seine Anwendbarkeit in chemisch-biologischen und auch medizinischen Studien geprüft.