Forschungsbericht 2005 - Max-Planck-Institut für Psychiatrie
Genetische Grundlagen der Wirksamkeit von Antidepressiva
Individuelle antidepressive Therapie
Seit der Einführung der Antidepressiva in den 1960er-Jahren ist bekannt, dass Patienten, die an einer Depression leiden, sehr unterschiedlich auf diese Medikamente reagieren. Während es bei manchen Betroffenen zu einer raschen Besserung kommt, bleibt diese bei anderen aus, sodass das Präparat gewechselt werden muss. Derzeit ist es noch nicht möglich, die Wirkung eines Antidepressivums im Einzelfall vorherzusagen. Eine Prognose wäre aber von großer Bedeutung, sowohl für die individuelle Lebensgestaltung des Patienten, als auch aus gesundheitsökonomischen Gründen, da mit dem längeren Verlauf einer depressiven Phase erhebliche Kosten verbunden sind. Hierbei ist zu bedenken, dass eine Entscheidung über die Notwendigkeit des Wechsels auf ein anderes Antidepressivum erst nach etwa vier Wochen Therapiedauer sinnvoll ist.
Trotz vielfältiger Bemühungen war es bisher nicht möglich, aufgrund von Unterschieden im klinischen Erscheinungsbild – wie etwa dem Ausprägungsmuster oder der Schwere der Symptome, der Häufigkeit von depressiven Episoden oder dem Krankheitsbeginn – vorherzusagen, wie der einzelne Patient auf ein bestimmtes Antidepressivum anspricht. Für die Wissenschaftler am MPI für Psychiatrie in München ist außerdem von Interesse, ob bestimmte genetische Varianten die Wahrscheinlichkeit des Auftretens und den Verlauf anderer komplexer Erkrankungen beeinflussen, zum Beispiel der Multiplen Sklerose, der Angsterkrankungen und des Restless Legs Syndroms, einer während des Schlafs auftretenden Bewegungsstörung.
Aus diesem Grunde wird am MPI für Psychiatrie untersucht, ob Veränderungen im Erbgut der Patienten solche Vorhersagen erlauben. Zu diesem Zweck war es zunächst erforderlich, große Patienten- und Kontrollkollektive zu rekrutieren, die aus mehreren tausend Personen bestehen und sehr umfassend phänotypisiert werden. Darunter versteht man die möglichst genaue Erfassung individueller biologischer und psychologischer Parameter.
Nachfolgend sollen die Möglichkeiten und Vorgehensweisen der Genetik für diese Fragestellungen dargestellt werden. Die Forschung wird in Zusammenarbeit mehrerer Arbeitsgruppen geleistet, insbesondere der Gruppen um Florian Holsboer, Marcus Ising, Manfred Uhr und Bertram Müller-Myhsok, aber auch der Arbeitsgruppen von Theo Rein, Christoph W. Turck, Frank Weber, Juliane Winkelmann und Jan Deussing.
Polymorphismen der DNS
Die grundlegende Tatsache, dass Teile der menschlichen Erbsubstanz (DNS) variabel (polymorph) sind, ist seit längerem bekannt. Unter den verschiedenen Klassen von Polymorphismen scheint der Austausch einzelner Basen (single nucleotide polymorphisms, SNPs) besonders bedeutsam zu sein, der in sehr großer Anzahl – mehrere Millionen Mal über das gesamte menschlichen Genom verteilt – vorkommt. Je nach Lage bezüglich eines Gens können SNPs etwa die Proteinsequenz des kodierten Eiweißes, die Aktivität des Gens oder die Modifikationen des Genprodukts durch Spleißen (Veränderung der Boten-RNA im Zellkern) beeinflussen. Manche SNPs liegen auch in den intronischen Teilen, den nicht kodierenden Abschnitten eines Gens, weshalb ihnen nicht ohne weiteres eine Funktion zugeordnet werden kann. Allerdings treten Polymorphismen meist in Abhängigkeit von anderen benachbarten Polymorphismen auf (Kopplungsungleichgewicht), sodass aus dem gehäuften Vorkommen eines Polymorphismus bei einer Gruppe von Patienten (Assoziation) folgt, dass dies auch auf benachbarte Polymorphismen zutrifft (Abb. 1). Die Genotypisierung der Proben erfolgt im Center for Applied Genotyping (CAGT), einer Einrichtung des Max-Planck Instituts für Psychiatrie in Zusammenarbeit mit dem GSF-Forschungszentrum für Umwelt und Gesundheit (Neuherberg). Hierbei stehen mehrere Hoch- und Höchstdurchsatztechnologien zur Genotypisierung zur Verfügung, wobei der bei weitem größte Durchsatz an genomweiten Analysen mit einer Anlage der Firma Illumina erreicht wird. Mit dieser Technologie werden derzeit mehr als sechs Millionen Genotypen pro Woche erzeugt. Eine weitere wesentliche Steigerung des Durchsatzes ist für die ersten Monate des Jahres 2006 geplant.

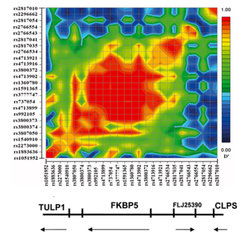
Assoziationsstudien
Assoziationsstudien können als eine Spezialform genetischer Kartierungsverfahren aufgefasst werden. Hierbei wird verglichen, ob sich innerhalb zweier Gruppen von Individuen, z. B. bei Patienten, die auf eine antidepressive Behandlung ansprechen, und solchen, bei denen dies nicht der Fall ist, die Häufigkeiten bestimmter genetischer Merkmale signifikant unterscheiden. Dabei kann prinzipiell festgestellt werden, ob sich die Häufigkeiten von SNPs unterscheiden. Dies erfolgt mithilfe diverser statistischer Verfahren; insbesondere kommen Verfahren zum Einsatz, die den Einfluss von sog. Kovariablen berücksichtigen können. Solche Kovariablen sind beispielweise die Verlaufsform der Erkrankung, das Alter, das Geschlecht, biochemische Parameter und Umweltfaktoren. Bei diesen Verfahren wird deutlich erkennbar, dass die Qualität der Phänotypisierung, also das Ausmaß der Kenntnis der Kovariablen, über den Erfolg oder Misserfolg einer Studie entscheidet. Deshalb ist für die humangenetischen Studien am MPI für Psychiatrie der unmittelbare Zugang zu den Patienten von größter Wichtigkeit.
Multiples Testen
Aufgrund der Vielzahl der zu untersuchenden Polymorphismen ist es notwendig, die Ergebnisse für die Zahl der durchgeführten Tests zu korrigieren. Dies ergibt sich aus der deutlich erhöhten Wahrscheinlichkeit, dass infolge der großen Menge von statistischen Tests rein zufällig viele Tests die gewählte Signifikanzschwelle überschreiten. Nimmt man als Signifikanzschwelle eine Wahrscheinlichkeit von 0.05 an, so bedeutet dies für einen einzelnen Test, dass dieser in einem von zwanzig Fällen diese Signifikanzschwelle unterschreitet. Bei zehn durchgeführten Tests ist die Wahrscheinlichkeit, dass mindestens einer der Tests diese Schwelle unterschreitet, bereits auf 40,1 Prozent erhöht, bei 100 Tests auf 99,4%. Daher muss der Zahl der durchgeführten Tests Rechnung getragen werden. Weiterhin bedingt die Korrelationsstruktur der Polymorphismen, dass Standardverfahren der Korrektur, wie etwa die Methoden nach Bonferroni oder Sídàk, die dies nicht berücksichtigen, zu einer Überkorrektur führen.
Daher werden Monte-Carlo-Verfahren bevorzugt, die durch Computersimulationen den Einfluss der internen Korrelationsstruktur der Polymorphismen auf das Verhalten und die Abhängigkeiten der statistischen Tests untereinander bestimmen und für die Korrektur berücksichtigen. Diese Verfahren sind sehr zeitaufwändig, da insbesondere bei vielen Polymorphismen und den geforderten hohen Signifikanzen (sog. p-Werte kleiner als 1/100000) meist mehrere Millionen Computersimulationen durchgeführt werden müssen. Aus diesen Gründen wurde am Max-Planck-Institut für Psychiatrie in der Arbeitsgruppe Statistische Genetik ein Verfahren entwickelt [1], das die benötigte Rechenzeit um den Faktor 100 reduzieren kann.
Mehrere Faktoren in einem Gen können relevant sein
Gelegentlich herrscht in der genetischen Forschung implizit noch das Mutationsparadigma vor, d. h. die Vorstellung, dass innerhalb eines Gens genau eine Mutation relevant für einen Phänotyp sei. Dies ist allerdings nur eine Modellvorstellung, die nicht immer zutreffen muss. Insbesondere können neben einer Vielzahl seltener Mutationen, wie sie etwa in der Krebsforschung für das p23-Gen beschrieben wurden, auch mehrere häufigere Polymorphismen unabhängig voneinander Effekte auf den gleichen Phänotyp bewirken.
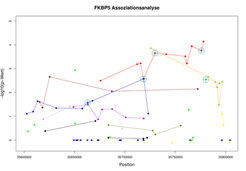
Anhand des FKBP5-Gens, das 2004 von Binder et al. [2] aus dem Max-Planck-Institut für Psychiatrie als Vorhersagefaktor für das Ansprechen von Patienten auf die Behandlung mit Antidepressiva beschrieben wurde (Abb. 2 und Abb. 3), lässt sich zeigen, dass durch eine Erhöhung der Dichte der untersuchten Polymorphismen (von 32 auf 91) auch die Auftrennung zunächst nicht unterscheidbarer Effekte gelingt (Abb. 4).
 nicht voneinander zu trennen waren.")

 zeigen ein deutlich schnelleres therapeutisches Ansprechen auf Antidepressiva, unabhängig von der Art der Medikation.")
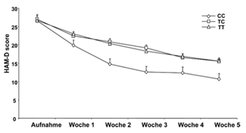
, deren Effekte nun (91 SNPs) deutlich voneinander zu trennen sind.")
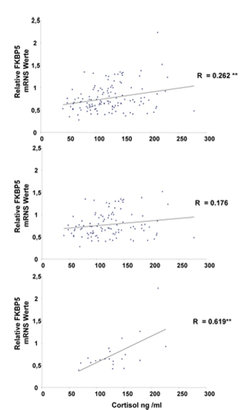
Interessanterweise beeinflussen die Polymorphismen nicht nur das Ansprechen auf Antidepressiva als solches, sondern auch die Produktion des FK506 binding protein 5 (FKBP5). Dieses Protein ist für die Regulation der Funktion von Corticosteroid-Rezeptoren wichtig, wie anhand des Polymorphismus rs1360780 gezeigt werden kann. Dieser Polymorphismus weist zwei Allele auf, bei denen entweder die Nukleinsäure Cytosin (C) oder Thymidin (T) in der DNS-Sequenz vorkommen. Das Vorliegen von zwei T-Allelen ist mit einer sehr viel höheren Wahrscheinlichkeit verbunden, unter antidepressiver Behandlung eine Besserung (Abb. 3) zu erfahren, außerdem mit einer erhöhten Produktion von FKBP5 (Abb. 5).

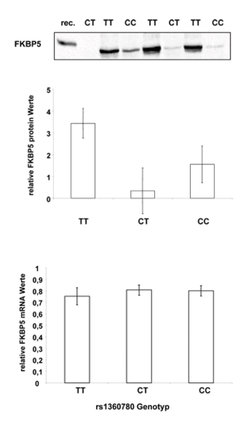
Darüber hinaus wirken sich die Polymorphismen auch auf den Zusammenhang zwischen dem FKBP5-Protein und der Serum-Cortisol-Konzentration aus (Abb. 6). Dies deutet darauf hin, dass der Polymorphismus (oder ein anderer, mit ihm in Kopplungsungleichgewicht stehender Polymorphismus) die Funktion des FKBP5-Proteins bezüglich der Corticosteroid-Rezeptorenfunktion moduliert, was eine biologische Erklärung für den Einfluss dieses Polymorphismus auf das Ansprechverhalten bei Antidepressiva liefert.

Ausblick und Zusammenfassung
Die beschriebenen Methoden der Assoziationsstudien lassen sich nicht nur auf einzelne Gene, sondern auch auf das gesamte menschliche Genom anwenden. Hierzu müssen zunächst mehrere hunderttausend Polymorphismen gleichzeitig im Labor bestimmt werden. Dies geschieht derzeit am CAGT des Max-Planck-Instituts für Psychiatrie. Aufgrund der hierdurch gegebenen Möglichkeit, sehr große Zahlen von Polymorphismen in einem Genom parallel zu analysieren, sind hypothesengeleitete Vorannahmen über die funktionelle bzw. physiologische Relevanz einzelner Gene nicht mehr erforderlich, die bisher infolge der methodischen Einschränkungen unumgänglich waren. Dieser ergebnisoffene Ansatz wird daher viele neue, möglicherweise überraschende und auch für die Anwendung relevante Befunde zeitigen.
Das Beispiel des FKBP5-Gens zeigt darüber hinaus auf, wie sich aus einer klinischen Fragestellung, nämlich der individuellen Prognose des Therapieverlaufs unter Antidepressiva, richtungweisende wissenschaftliche Fragestellungen in der statistischen Genetik (neue Analyseverfahren), der Bioinformatik (verteiltes Rechnen, schnellere Algorithmen) und der funktionellen Genomik (Hochdurchsatz-Methoden) ergeben, die alle derzeit am Max-Planck-Institut für Psychiatrie bearbeitet werden.