Forschungsbericht 2005 - Max-Planck-Institut für Biochemie
Biochemie und Zellbiologie Proteolyse-vermittelnder Proteinkomplexe
Proteolysis-mediating Protein Complexes
Autoren
Buchberger, Alexander
Abteilungen
Biochemie und Zellbiologie Proteolyse-vermittelnder Proteinkomplexe (Buchberger, Abteilung Molekulare Zellbiologie, DFG/Emmy Noether) (Dr. Alexander Buchberger) MPI für Biochemie, Martinsried
Zusammenfassung
Untersucht wird die Regulation der Substratspezifität im Ubiquitin-Proteasom-System. Der Forschungsschwerpunkt liegt dabei auf zwei modular aufgebauten Proteinkomplexen: der CBCVHL Ubiquitin-Ligase mit dem von-Hippel-Lindau Tumorsuppressor-Protein als substratbindender Untereinheit und der Chaperon-ähnlichen AAA ATPase Cdc48 mit den Kofaktoren der UBX-Proteinfamilie. UBX-Proteine binden an Cdc48 und regulieren so die Spezifität der Cdc48-Aktivität in verschiedenen zellulären Prozessen. UBX-Proteine mit einer Ubiquitin-bindenden UBA-Domäne rekrutieren ubiquitylierte Substrate, die durch Cdc48 der proteasomalen Degradation zugeführt werden. Ein solches UBA/UBX-Protein, Ubx2 genannt, spielt eine zentrale Rolle in der Endoplasmatischen Retikulum (ER) assoziierten Proteindegradation (ERAD). Biochemische Untersuchungen von Tumor-assoziierten Mutanten des von-Hippel-Lindau Tumorsuppressor-Proteins erlauben neue Einblicke in die Genotyp/Phänotyp-Beziehungen der von-Hippel-Lindau´schen Erbkrankheit. So korreliert der Grad der funktionellen Defekte auf molekularer Ebene mit dem Risiko der Patienten, Nierenzellkarzinome zu entwickeln.
Summary
Our research focuses on the regulation of substrate specificity in the ubiquitin proteasome system, specifically on two modular protein complexes: the CBCVHL ubiquitin ligase with its substrate binding subunit, the von-Hippel-Lindau tumor suppressor protein, and the chaperone-like Cdc48 AAA ATPase with cofactors of the UBX protein family. UBX proteins bind to Cdc48 and thereby regulate the specificity of Cdc48 activity in various cellular processes. UBX proteins with a ubiquitin binding UBA domain recruit ubiquitylated substrates which are targeted via Cdc48 for proteasomal degradation. One such UBA/UBX protein, called Ubx2, plays a central role in endoplasmic reticulum (ER) associated protein degradation (ERAD). Biochemical studies of tumor associated mutants of the von-Hippel-Lindau tumor suppressor protein provide new insights in the complex genotype/phenotype relationship of the von-Hippel-Lindau disease. For instance, the extent of functional defects on the molecular level correlates with the patients´ risk of developing renal cell carcinomas.
Das Ubiquitin-Proteasom-System
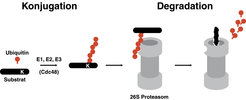
Das Ubiquitin-Proteasom-System für den selektiven Abbau von Proteinen ist in allen eukaryotischen Organismen in konservierter Form vorhanden und spielt eine zentrale Rolle in einer Vielzahl von lebenswichtigen zellulären Prozessen wie Zellzyklus, Qualitätskontrolle der Proteinfaltung, Signaltransduktion, Genexpression und Apoptose (programmierter Zelltod). Substratproteine werden durch Ubiquitin kovalent modifiziert und so für den prozessiven Abbau durch das 26S Proteasom markiert (Abb. 1).
Das Ubiquitin-Proteasom-System. Die kovalente Bindung von Ubiquitin (rot) mit einer Lysin-Seitenkette (K) des Substratproteins („Ubiquitylierung”) benötigt die Enzyme E1, E2, E3 sowie bei manchen Substraten die AAA ATPase Cdc48. Die Ubiquitylierung von Lysinresten in Ubiquitin selbst führt zur Bildung einer Ubiquitinkette, die das Substratprotein für den proteolytischen Abbau durch das 26S Proteasom markiert. Die Ubiquitinkette wird am 26S Proteasom durch spezifische Proteasen wieder in einzelne Ubiquitinbausteine zerlegt, die erneut verwendet werden können.
Das Ubiquitin-Proteasom-System. Die kovalente Bindung von Ubiquitin (rot) mit einer Lysin-Seitenkette (K) des Substratproteins („Ubiquitylierung”) benötigt die Enzyme E1, E2, E3 sowie bei manchen Substraten die AAA ATPase Cdc48. Die Ubiquitylierung von Lysinresten in Ubiquitin selbst führt zur Bildung einer Ubiquitinkette, die das Substratprotein für den proteolytischen Abbau durch das 26S Proteasom markiert. Die Ubiquitinkette wird am 26S Proteasom durch spezifische Proteasen wieder in einzelne Ubiquitinbausteine zerlegt, die erneut verwendet werden können.
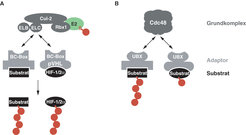
Da das 26S Proteasom Substrate nicht anhand intrinsischer Merkmale, sondern ausschließlich aufgrund ihrer kovalenten Markierung mit Ubiquitin abbaut, ist die Regulation der Substratspezifität für das Ubiquitin-Proteasom-System von zentraler Bedeutung. Die Ubiquitylierung von Substratproteinen erfolgt über eine Kaskade von drei enzymatischen Aktivitäten, dem Ubiquitin-aktivierenden Enzym (E1), einem Ubiquitin-konjugierenden Enzym (E2) und einer Ubiquitin-Ligase (E3). Ubiquitin-Ligasen bilden eine heterogene Klasse von Proteinen, deren gemeinsame Funktion die Erkennung von spezifischen Degradationssignalen in Substratproteinen ist. Ubiquitin-Ligasen sind somit maßgeblich an der Substratspezifität des Ubiquitin-Proteasom-Systems beteiligt. Dabei zeichnet sich die Familie der heterooligomeren, Cullin-enthaltenden Ubiquitin-Ligasen durch eine besonders große Substratdiversität aus: Unterschiedliche, modular aufgebaute Adaptorproteine erkennen spezifische Substrate und rekrutieren diese an einen invarianten, enzymatisch aktiven Grundkomplex (Abb. 2A). Ein Vertreter dieser Familie mit besonderer medizinischer Bedeutung ist der CBCVHL Ubiquitin-Ligase-Komplex, dessen Adaptoruntereinheit das von-Hippel-Lindau Tumorsuppressor-Protein ist.
Modulare Adaptorproteine als Spezifitätsfaktoren im Ubiquitin-Proteasom-System. (A) Der Cullin-2 Ubiquitin-Ligase-Komplex. BC-Box Proteine (hellgrau) rekrutieren spezifische Substrate (schwarz) an den konstitutiven Grundkomplex (dunkelgrau) der CBC Ubiquitin-Ligase-Familie. Dieser bindet ein Ubiquitin-konjugierendes Enzym (E2; grün), das aktiviertes Ubiquitin (rot) auf das Substratprotein überträgt. Das von-Hippel-Lindau Tumorsuppressorprotein (pVHL) ist der Substratadaptor des CBCVHL Ubiquitin-Ligase-Komplexes und vermittelt dessen Substratspezifität für die Transkriptionsfaktoren HIF-1/2 α. (B) Cdc48. UBX-Domänen-Proteine (hellgrau) rekrutieren spezifische ubiquitylierte Substrate (schwarz) an die hexamere, Chaperon-ähnliche AAA ATPase Cdc48 (dunkelgrau).
Modulare Adaptorproteine als Spezifitätsfaktoren im Ubiquitin-Proteasom-System. (A) Der Cullin-2 Ubiquitin-Ligase-Komplex. BC-Box Proteine (hellgrau) rekrutieren spezifische Substrate (schwarz) an den konstitutiven Grundkomplex (dunkelgrau) der CBC Ubiquitin-Ligase-Familie. Dieser bindet ein Ubiquitin-konjugierendes Enzym (E2; grün), das aktiviertes Ubiquitin (rot) auf das Substratprotein überträgt. Das von-Hippel-Lindau Tumorsuppressorprotein (pVHL) ist der Substratadaptor des CBCVHL Ubiquitin-Ligase-Komplexes und vermittelt dessen Substratspezifität für die Transkriptionsfaktoren HIF-1/2 α. (B) Cdc48. UBX-Domänen-Proteine (hellgrau) rekrutieren spezifische ubiquitylierte Substrate (schwarz) an die hexamere, Chaperon-ähnliche AAA ATPase Cdc48 (dunkelgrau).
Für die Degradation vieler Substrate des Ubiquitin-Proteasom-Systems werden nicht nur spezifische Ubiquitin-Ligasen benötigt, sondern auch Cdc48, ein homohexamerer, Chaperon-ähnlicher Komplex aus der Familie der AAA ATPasen. Die Funktion von Cdc48 scheint darin zu bestehen, Energie aus der Hydrolyse von ATP in mechanische Energie umzuwandeln, um ubiquitylierte Proteine zu entfalten oder aus stabilen Proteinkomplexen herauszulösen. Analog zu den Ubiquitin-Ligasen der CBC-Familie wird auch die Substratspezifität von Cdc48 durch Adaptorproteine, darunter die Gruppe der UBX-Domänen enthaltenden Proteine, reguliert (Abb. 2B).
Unser wissenschaftliches Interesse gilt der Regulation der Substratspezifität im Ubiquitin-Proteasom-System. Die Forschung konzentriert sich dabei auf den CBCVHL Ubiquitin-Ligase-Komplex und den Chaperon-ähnlichen Cdc48-Komplex mit dem Ziel, die Funktion der Spezifität vermittelnden Adaptoruntereinheiten auf molekularer und zellulärer Ebene zu verstehen.
UBX-Proteine bilden eine neue Familie von Cdc48-Kofaktoren
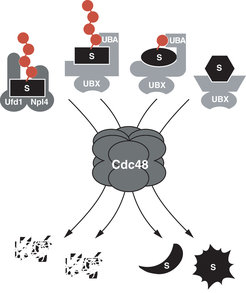
Cdc48 (in Säugern als p97 oder VCP bezeichnet) ist eine Chaperon-ähnliche molekulare Maschine, die ubiquitylierte Substrate bindet und entweder der proteasomalen Degradation zuführt oder für nicht-degradative Prozesse freisetzt. Die unterschiedlichen zellulären Aktivitäten von Cdc48 werden dabei durch distinkte Adaptoren vermittelt (Abb. 3). So rekrutiert der heterodimere Kofaktor Ufd1/Npl4 Substrate aus verschiedenen proteasomalen Abbauwegen an Cdc48, darunter im Rahmen der zellulären Qualitätskontrolle auch fehlgefaltete Proteine aus Lumen und Membran des endoplasmatischen Retikulums (ER). Dagegen ist Shp1 (p47 in Säugern) essenziell für die Funktion von Cdc48 bei der Fusion homotypischer Membranen des ER und Golgi Apparates. Diese Funktion von Cdc48 ist zwar unabhängig von proteasomaler Degradation, involviert aber ebenfalls ubiquitylierte Substrate.
Substrat-rekrutierende Kofaktoren von Cdc48. Cdc48 bindet präferenziell Substrate (schwarz; S), die durch eine oder mehrere Ubiquitinreste (rot) kovalent markiert sind. Multiubiquitylierte Substrate werden durch den heterodimeren Kofaktor Ufd1/Npl4 und/oder durch UBA/UBX-Domänenproteine rekrutiert und der proteasomalen Degradation zugeführt. Monoubiquitylierte Substrate werden ebenfalls von UBA/UBX-Domänenproteinen (z.B. Shp1) rekrutiert, aber als stabile Proteine wieder freigesetzt. Die Rekrutierung nicht-ubiquitylierter Substrate durch UBX-Domänenproteine (rechts) ist hypothetisch.
Substrat-rekrutierende Kofaktoren von Cdc48. Cdc48 bindet präferenziell Substrate (schwarz; S), die durch eine oder mehrere Ubiquitinreste (rot) kovalent markiert sind. Multiubiquitylierte Substrate werden durch den heterodimeren Kofaktor Ufd1/Npl4 und/oder durch UBA/UBX-Domänenproteine rekrutiert und der proteasomalen Degradation zugeführt. Monoubiquitylierte Substrate werden ebenfalls von UBA/UBX-Domänenproteinen (z.B. Shp1) rekrutiert, aber als stabile Proteine wieder freigesetzt. Die Rekrutierung nicht-ubiquitylierter Substrate durch UBX-Domänenproteine (rechts) ist hypothetisch.
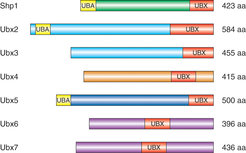
Shp1 ist ein Cdc48-Kofaktor aus der Familie der UBX-Proteine. Diese von der Bäckerhefe Saccharomyces cerevisiae bis zum Menschen evolutionär konservierte Familie ist durch das Vorhandensein der Ubiquitin ähnlichen UBX-Domäne definiert [1; 2]. Der Modellorganismus S. cerevisiae besitzt sieben UBX-Proteine, die Homologe in höheren Eukaryoten haben (Abb. 4). Wir konnten zeigen, dass alle sieben UBX-Proteine an Cdc48 binden und dass die UBX-Domäne ein allgemeines Cdc48-Bindemodul ist [3]. Dies lässt vermuten, dass neben Shp1 auch andere UBX-Proteine Adaptoren für Substrate von Cdc48 sind (vgl. Abb. 3). Im Gegensatz zur gut etablierten Funktion von Shp1 in Membranfusionsprozessen sind die meisten anderen Vertreter dieser Familie jedoch nicht näher charakterisiert. Unser Ziel ist es daher, die spezifischen zellulären Substrate und Prozesse, an denen UBX-Proteine beteiligt sind, zu identifizieren.
UBX-Proteine der Bäckerhefe Saccharomyces cerevisiae. Die UBX-Proteine wurden aufgrund ihrer Sequenzhomologie außerhalb der UBX-Domäne (rot) gruppiert und farblich unterschieden. Die Ubiquitin-bindende UBA-Domäne von Shp1, Ubx2 und Ubx5 ist gelb markiert.
UBX-Proteine der Bäckerhefe Saccharomyces cerevisiae. Die UBX-Proteine wurden aufgrund ihrer Sequenzhomologie außerhalb der UBX-Domäne (rot) gruppiert und farblich unterschieden. Die Ubiquitin-bindende UBA-Domäne von Shp1, Ubx2 und Ubx5 ist gelb markiert.
UBA/UBX-Proteine rekrutieren ubiquitylierte Substrate an Cdc48
Eine Untergruppe von UBX-Proteinen besitzt eine aminoterminale UBA-Domäne (vgl. Abb. 4), ein Bindemodul für Ubiquitinketten und ubiquitylierte Substrate. In der Tat binden die drei UBA/UBX-Proteine aus der Bäckerhefe, Shp1, Ubx2 und Ubx5, ubiquitylierte Proteine in vivo [3]. Wir konnten zeigen, dass Shp1 und Ubx2 an der proteasomalen Degradation des Modellsubstrates Ubiquitin-Pro-β-Galaktosidase beteiligt sind. Darüber hinaus sind Hefestämme ohne Shp1 bzw. Ubx2 (shp1Δ und ubx2Δ Stämme) hypersensitiv gegenüber denjenigen Stressbedingungen, die zur Akkumulation fehlgefalteter Proteine führen. Diese Ergebnisse legen eine Funktion von Shp1 und Ubx2 als Substrat-rekrutierende Adaptoren in der Cdc48-vermittelten zellulären Protein-Qualitätskontrolle nahe - eine Rolle, die auch für Shp1 bisher unbekannt war [3].
Ubx2 ist eine wichtige Komponente im ERAD-Abbauweg
In weiteren Studien konnte die zelluläre Funktion von Ubx2 als zentraler Faktor in der ER-assoziierten Proteindegradation (ERAD) aufgeklärt werden [4]. ERAD ist ein wichtiger Bestandteil der zellulären Proteinqualitätskontrolle. In diesem Abbauweg werden fehlgefaltete Proteine des ER durch eine Proteinpore in der ER-Membran (Retrotranslokationspore) zurück in das Zytosol transportiert und anschließend der proteasomalen Degradation zugeführt. Der Cdc48Ufd1/Npl4-Komplex ist essenziell für den Rücktransport von ERAD-Substraten. Ubx2 ist ein integrales Membranprotein des ER und wirkt über seine UBX-Domäne als Membrananker für den Cdc48Ufd1/Npl4-Komplex (Abb. 5A). Darüber hinaus interagiert Ubx2 mit ERAD-Substraten, deren spezifischen Ubiquitin-Ligasen und postulierten Komponenten der Retrotranslokationspore und wird für die stabile Assoziation von Cdc48Ufd1/Npl4 mit allen diesen Proteinen benötigt (Abb. 5B). Das Fehlen von Ubx2 verursacht Defekte im ERAD Abbauweg, welche unter Stressbedingungen, die zur Akkumulation fehlgefalteter Proteine im ER führen, besonders ausgeprägt sind [4]. Die Untersuchung der zahlreichen anderen UBX-Proteine in Hefe und in höheren Eukaryoten verspricht weitere interessante Einblicke in die Regulation der Substratspezifität von Cdc48.
Rolle von Ubx2 in der ER-assoziierten Proteindegradation (ERAD). (A) Lebendzell-Fluoreszenzmikroskopie von Hefezellen. GFP (green fluorescent protein)-markiertes Ubx2 (grün) ist im perinukleären und kortikalen ER lokalisiert. (B) Das integrale ER-Membranprotein Ubx2 koordiniert multiple Interaktionen entlang des ERAD-Abbauweges. ERAD-Substrat (schwarz; S), das auf der zytosolischen Seite der Retrotranslokationspore (?; Der1? Dfm1?) erscheint, wird durch eine ERAD-spezifische Ubiquitin-Ligase (E3) erkannt und ubiquityliert (helle Kreise). Das ubiquitylierte Substrat wird durch den Cdc48Ufd1/Npl4-Komplex komplett in das Zytosol transportiert und zur Degradation an das 26S Proteasom weitergeleitet. Ubx2 interagiert mit Substrat und allen Proteinen entlang dieses Abbauweges und rekrutiert über seine UBX-Domäne Cdc48Ufd1/Npl4 [4].
Rolle von Ubx2 in der ER-assoziierten Proteindegradation (ERAD). (A) Lebendzell-Fluoreszenzmikroskopie von Hefezellen. GFP (green fluorescent protein)-markiertes Ubx2 (grün) ist im perinukleären und kortikalen ER lokalisiert. (B) Das integrale ER-Membranprotein Ubx2 koordiniert multiple Interaktionen entlang des ERAD-Abbauweges. ERAD-Substrat (schwarz; S), das auf der zytosolischen Seite der Retrotranslokationspore (?; Der1? Dfm1?) erscheint, wird durch eine ERAD-spezifische Ubiquitin-Ligase (E3) erkannt und ubiquityliert (helle Kreise). Das ubiquitylierte Substrat wird durch den Cdc48Ufd1/Npl4-Komplex komplett in das Zytosol transportiert und zur Degradation an das 26S Proteasom weitergeleitet. Ubx2 interagiert mit Substrat und allen Proteinen entlang dieses Abbauweges und rekrutiert über seine UBX-Domäne Cdc48Ufd1/Npl4 [4].
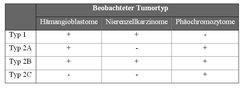
Das von-Hippel-Lindau Tumorsupressor-Protein (pVHL) ist die Substrat erkennende Untereinheit des CBCVHL Ubiquitin-Ligase-Komplexes. Mutationen im VHL Gen beeinträchtigen die Funktion von CBCVHL und verursachen die von Hippel-Lindau´sche Erbkrankheit, die sich in verschiedenen benignen und malignen Tumoren manifestiert (Abb. 6). Bei Typ 1 treten Hämangioblastome in Retina und zentralem Nervensystem (ZNS) sowie klarzellige Nierenzellkarzinome auf, nicht aber Phäochromozytome der Nebennierenrinde. Typ 2 ist definiert durch das Auftreten von Phäochromozytomen, entweder als alleiniges Krankheitsmerkmal (Typ 2C), zusammen mit Hämangioblastomen (Typ 2A), oder zusammen mit Hämangioblastomen und Nierenzellkarzinomen (Typ 2B). Darüber hinaus ist das VHL Gen auch in der Mehrzahl der spontan auftretenden, klarzelligen Nierenzellkarzinome mutiert.
Klinische Einteilung der von Hippel-Lindau´schen Krankheit. Grundlage für die Einteilung in verschiedene Krankheitstypen ist das unterschiedliche Auftreten von Hämangioblastomen, klarzelligen Nierenzellkarzinomen und Phäochromozytomen. (+) hohes Risiko; (-) kein oder geringes Risiko.
Klinische Einteilung der von Hippel-Lindau´schen Krankheit. Grundlage für die Einteilung in verschiedene Krankheitstypen ist das unterschiedliche Auftreten von Hämangioblastomen, klarzelligen Nierenzellkarzinomen und Phäochromozytomen. (+) hohes Risiko; (-) kein oder geringes Risiko.
Das Schlüsselsubstrat der CBCVHL Ubiquitin-Ligase ist HIF 1α, die alpha-Untereinheit des Hypoxia-induzierten Transkriptionsfaktors 1. Während HIF 1α unter normoxischen Bedingungen (bei normaler Sauerstoffkonzentration) von CBCVHL ubiquityliert und rasch durch das 26S Proteasom abgebaut wird, ist das Protein unter hypoxischen Bedingungen (bei Sauerstoffmangel) aufgrund des Fehlens einer für die Erkennung durch CBCVHL kritischen, posttranslationalen Modifikation stabil. HIF 1α bildet dann mit HIF 1β (ARNT) einen aktiven Transkriptionsfaktor für die Expression von Genen, die die Nährstoffversorgung und Vaskularisierung von Geweben steuern. Viele der im Zusammenhang mit der von Hippel-Lindau´schen Krankheit identifizierten VHL Mutationen führen zum Verlust der Rekrutierung von HIF 1α an CBCVHL und somit zur konstitutiven Aktivierung HIF 1-abhängiger Gene auch unter normoxischen Bedingungen. Allerdings sind die molekularen Konsequenzen von Tumor-assoziierten VHL-Mutationen und insbesondere das Verhältnis zwischen Genotyp (Mutation) und Phänotyp (Krankheitstyp) nicht im Detail verstanden. Unser Ziel ist es daher, die funktionellen Auswirkungen Tumor-assoziierter VHL-Mutationen auf molekularer Ebene aufzuklären. Insbesondere untersuchen wir, warum unterschiedliche Mutationen mit verschiedenen Krankheitstypen assoziiert sind.
Nierenzellkarzinome
Die Analyse Tumor-assoziierter VHL Mutationen wurde zunächst auf zwei Paare von Mutanten fokussiert, bei denen unterschiedliche Austausche derselben Aminosäure zu unterschiedlichen Krankheitstypen führen [5] (Abb. 7). Die Aminosäure-Austausche Y98H und Y112H wurden in Patienten mit Krankheitstyp 2A (geringe Wahrscheinlichkeit von Nierenzellkarzinomen) identifiziert, während die Austausche Y98N und Y112N mit Krankheitstyp 2B (hohe Wahrscheinlichkeit von Nierenzellkarzinomen) korreliert werden konnten. Alle Mutantenproteine assemblierten wie Wildtyp pVHL in CBCVHL Ubiquitin-Ligase Komplexe, zeigten aber im Vergleich zum Wildtyp reduzierte Stabilität, deutlich schlechtere Substratbindung und niedrigere Ubiquitin-Ligase Aktivität gegenüber HIF 1α. Dabei wiesen die Mutantenproteine Y98H und Y112H jeweils geringere Defekte als Y98N und Y112N auf. Insbesondere wurden Y98H und Y112H, aber nicht Y98N und Y112N bei der physiologisch relevanten Temperatur von 37 oC durch Substratbindung stabilisiert und besaßen signifikante Restaktivität in ihrer Funktion als Ubiquitin-Ligase. Die mit Krankheitstyp 2A assoziierten Mutanten sind funktionell demnach weniger beeinträchtigt als die des Typs 2B, so dass ein Zusammenhang zwischen dem Grad der funktionellen Defekte auf molekularer Ebene und der Schwere des Krankheitsverlaufes nachgewiesen werden konnte [5].
Mit den in unserer Gruppe etablierten biochemischen Tests werden derzeit weitere Tumor-assoziierte Mutationen in VHL quantitativ analysiert, um auch Einblicke in die molekulare Grundlage des Krankheitstyps 2C zu erhalten. Darüber hinaus verfolgen wir mit biochemischen und genetischen Ansätzen das Ziel, weitere für die von Hippel-Lindau´sche Erbkrankheit kritische zelluläre Substrate der CBCVHL Ubiquitin Ligase zu identifizieren.
Struktur des von-Hippel-Lindau Tumorsuppressor-Proteins. (A) Dreidimensionale Struktur des pVHL/Elongin C/Elongin B/HIF-1α-Komplexes (PDB Eintrag 1LM8). Elongin C (ELC) und Elongin B (ELB) sind grau dargestellt, pVHL orange und ein von HIF-1α abgeleitetes Substratpeptid (Reste 561-575) grün. (B) Detailansicht der HIF-1α-Bindestelle nach Rotation von pVHL um etwa 180° um die vertikale Achse. Der für die Erkennung durch pVHL kritische Hydroxyprolinrest Hyp564 des Peptides ist gelb markiert. Die Seitenketten von Y98 und Y112 sind lila markiert. Beide Aminosäurereste sind an der Bindung des Substratpeptides beteiligt. Mutationen von Y98 und Y112 zu Asparagin (N) führen zu starken Defekten in der Substratbindung und –ubiquitylierung und sind mit einem hohen Risiko von Nierenzell-Karzinomen korreliert (Krankheitstyp 2B). Mutationen zu Histidin (H) verursachen weniger starke Defekte und sind mit einem niedrigen Risiko von Nierenzellkarzinomen korreliert (Krankheitstyp 2A) [5].
Struktur des von-Hippel-Lindau Tumorsuppressor-Proteins. (A) Dreidimensionale Struktur des pVHL/Elongin C/Elongin B/HIF-1α-Komplexes (PDB Eintrag 1LM8). Elongin C (ELC) und Elongin B (ELB) sind grau dargestellt, pVHL orange und ein von HIF-1α abgeleitetes Substratpeptid (Reste 561-575) grün. (B) Detailansicht der HIF-1α-Bindestelle nach Rotation von pVHL um etwa 180° um die vertikale Achse. Der für die Erkennung durch pVHL kritische Hydroxyprolinrest Hyp564 des Peptides ist gelb markiert. Die Seitenketten von Y98 und Y112 sind lila markiert. Beide Aminosäurereste sind an der Bindung des Substratpeptides beteiligt. Mutationen von Y98 und Y112 zu Asparagin (N) führen zu starken Defekten in der Substratbindung und –ubiquitylierung und sind mit einem hohen Risiko von Nierenzell-Karzinomen korreliert (Krankheitstyp 2B). Mutationen zu Histidin (H) verursachen weniger starke Defekte und sind mit einem niedrigen Risiko von Nierenzellkarzinomen korreliert (Krankheitstyp 2A) [5].
 mit einer Lysin-Seitenkette (K) des Substratproteins („Ubiquitylierung”) benötigt die Enzyme E1, E2, E3 sowie bei manchen Substraten die AAA ATPase Cdc48. Die Ubiquitylierung von Lysinresten in Ubiquitin selbst führt zur Bildung einer Ubiquitinkette, die das Substratprotein für den proteolytischen Abbau durch das 26S Proteasom markiert. Die Ubiquitinkette wird am 26S Proteasom durch spezifische Proteasen wieder in einzelne Ubiquitinbausteine zerlegt, die erneut verwendet werden können.")
 Der Cullin-2 Ubiquitin-Ligase-Komplex. BC-Box Proteine (hellgrau) rekrutieren spezifische Substrate (schwarz) an den konstitutiven Grundkomplex (dunkelgrau) der CBC Ubiquitin-Ligase-Familie. Dieser bindet ein Ubiquitin-konjugierendes Enzym (E2; grün), das aktiviertes Ubiquitin (rot) auf das Substratprotein überträgt. Das von-Hippel-Lindau Tumorsuppressorprotein (pVHL) ist der Substratadaptor des CBCVHL Ubiquitin-Ligase-Komplexes und vermittelt dessen Substratspezifität für die Transkriptionsfaktoren HIF-1/2 α. (B) Cdc48. UBX-Domänen-Proteine (hellgrau) rekrutieren spezifische ubiquitylierte Substrate (schwarz) an die hexamere, Chaperon-ähnliche AAA ATPase Cdc48 (dunkelgrau).")
, die durch eine oder mehrere Ubiquitinreste (rot) kovalent markiert sind. Multiubiquitylierte Substrate werden durch den heterodimeren Kofaktor Ufd1/Npl4 und/oder durch UBA/UBX-Domänenproteine rekrutiert und der proteasomalen Degradation zugeführt. Monoubiquitylierte Substrate werden ebenfalls von UBA/UBX-Domänenproteinen (z.B. Shp1) rekrutiert, aber als stabile Proteine wieder freigesetzt. Die Rekrutierung nicht-ubiquitylierter Substrate durch UBX-Domänenproteine (rechts) ist hypothetisch.")
 gruppiert und farblich unterschieden. Die Ubiquitin-bindende UBA-Domäne von Shp1, Ubx2 und Ubx5 ist gelb markiert.")
![Rolle von Ubx2 in der ER-assoziierten Proteindegradation (ERAD). (A) Lebendzell-Fluoreszenzmikroskopie von Hefezellen. GFP (green fluorescent protein)-markiertes Ubx2 (grün) ist im perinukleären und kortikalen ER lokalisiert. (B) Das integrale ER-Membranprotein Ubx2 koordiniert multiple Interaktionen entlang des ERAD-Abbauweges. ERAD-Substrat (schwarz; S), das auf der zytosolischen Seite der Retrotranslokationspore (?; Der1? Dfm1?) erscheint, wird durch eine ERAD-spezifische Ubiquitin-Ligase (E3) erkannt und ubiquityliert (helle Kreise). Das ubiquitylierte Substrat wird durch den Cdc48Ufd1/Npl4-Komplex komplett in das Zytosol transportiert und zur Degradation an das 26S Proteasom weitergeleitet. Ubx2 interagiert mit Substrat und allen Proteinen entlang dieses Abbauweges und rekrutiert über seine UBX-Domäne Cdc48Ufd1/Npl4 [4].](/333345/original-1293750192.jpg?t=eyJ3aWR0aCI6MzQxLCJmaWxlX2V4dGVuc2lvbiI6ImpwZyIsIm9ial9pZCI6MzMzMzQ1fQ%3D%3D--b2efe9ae28b5268e27497df4834cbbe10346f70d "Rolle von Ubx2 in der ER-assoziierten Proteindegradation (ERAD). (A) Lebendzell-Fluoreszenzmikroskopie von Hefezellen. GFP (green fluorescent protein)-markiertes Ubx2 (grün) ist im perinukleären und kortikalen ER lokalisiert. (B) Das integrale ER-Membranprotein Ubx2 koordiniert multiple Interaktionen entlang des ERAD-Abbauweges. ERAD-Substrat (schwarz; S), das auf der zytosolischen Seite der Retrotranslokationspore (?; Der1? Dfm1?) erscheint, wird durch eine ERAD-spezifische Ubiquitin-Ligase (E3) erkannt und ubiquityliert (helle Kreise). Das ubiquitylierte Substrat wird durch den Cdc48Ufd1/Npl4-Komplex komplett in das Zytosol transportiert und zur Degradation an das 26S Proteasom weitergeleitet. Ubx2 interagiert mit Substrat und allen Proteinen entlang dieses Abbauweges und rekrutiert über seine UBX-Domäne Cdc48Ufd1/Npl4 [4].")
![Rolle von Ubx2 in der ER-assoziierten Proteindegradation (ERAD). (A) Lebendzell-Fluoreszenzmikroskopie von Hefezellen. GFP (green fluorescent protein)-markiertes Ubx2 (grün) ist im perinukleären und kortikalen ER lokalisiert. (B) Das integrale ER-Membranprotein Ubx2 koordiniert multiple Interaktionen entlang des ERAD-Abbauweges. ERAD-Substrat (schwarz; S), das auf der zytosolischen Seite der Retrotranslokationspore (?; Der1? Dfm1?) erscheint, wird durch eine ERAD-spezifische Ubiquitin-Ligase (E3) erkannt und ubiquityliert (helle Kreise). Das ubiquitylierte Substrat wird durch den Cdc48Ufd1/Npl4-Komplex komplett in das Zytosol transportiert und zur Degradation an das 26S Proteasom weitergeleitet. Ubx2 interagiert mit Substrat und allen Proteinen entlang dieses Abbauweges und rekrutiert über seine UBX-Domäne Cdc48Ufd1/Npl4 [4].](/333345/original-1293750192.jpg?t=eyJ3aWR0aCI6MjQ2LCJvYmpfaWQiOjMzMzM0NX0%3D--6f836642b058fb5b2b4f3ef3d56e2f92aedf3ca7)
 hohes Risiko; (-) kein oder geringes Risiko.")
![Struktur des von-Hippel-Lindau Tumorsuppressor-Proteins. (A) Dreidimensionale Struktur des pVHL/Elongin C/Elongin B/HIF-1α-Komplexes (PDB Eintrag 1LM8). Elongin C (ELC) und Elongin B (ELB) sind grau dargestellt, pVHL orange und ein von HIF-1α abgeleitetes Substratpeptid (Reste 561-575) grün. (B) Detailansicht der HIF-1α-Bindestelle nach Rotation von pVHL um etwa 180° um die vertikale Achse. Der für die Erkennung durch pVHL kritische Hydroxyprolinrest Hyp564 des Peptides ist gelb markiert. Die Seitenketten von Y98 und Y112 sind lila markiert. Beide Aminosäurereste sind an der Bindung des Substratpeptides beteiligt. Mutationen von Y98 und Y112 zu Asparagin (N) führen zu starken Defekten in der Substratbindung und –ubiquitylierung und sind mit einem hohen Risiko von Nierenzell-Karzinomen korreliert (Krankheitstyp 2B). Mutationen zu Histidin (H) verursachen weniger starke Defekte und sind mit einem niedrigen Risiko von Nierenzellkarzinomen korreliert (Krankheitstyp 2A) [5].](/333457/original-1293750029.jpg?t=eyJ3aWR0aCI6MzQxLCJmaWxlX2V4dGVuc2lvbiI6ImpwZyIsIm9ial9pZCI6MzMzNDU3fQ%3D%3D--7079b387f61640dc5ab9fb3b230c96bdfd920f1d "Struktur des von-Hippel-Lindau Tumorsuppressor-Proteins. (A) Dreidimensionale Struktur des pVHL/Elongin C/Elongin B/HIF-1α-Komplexes (PDB Eintrag 1LM8). Elongin C (ELC) und Elongin B (ELB) sind grau dargestellt, pVHL orange und ein von HIF-1α abgeleitetes Substratpeptid (Reste 561-575) grün. (B) Detailansicht der HIF-1α-Bindestelle nach Rotation von pVHL um etwa 180° um die vertikale Achse. Der für die Erkennung durch pVHL kritische Hydroxyprolinrest Hyp564 des Peptides ist gelb markiert. Die Seitenketten von Y98 und Y112 sind lila markiert. Beide Aminosäurereste sind an der Bindung des Substratpeptides beteiligt. Mutationen von Y98 und Y112 zu Asparagin (N) führen zu starken Defekten in der Substratbindung und –ubiquitylierung und sind mit einem hohen Risiko von Nierenzell-Karzinomen korreliert (Krankheitstyp 2B). Mutationen zu Histidin (H) verursachen weniger starke Defekte und sind mit einem niedrigen Risiko von Nierenzellkarzinomen korreliert (Krankheitstyp 2A) [5].")
![Struktur des von-Hippel-Lindau Tumorsuppressor-Proteins. (A) Dreidimensionale Struktur des pVHL/Elongin C/Elongin B/HIF-1α-Komplexes (PDB Eintrag 1LM8). Elongin C (ELC) und Elongin B (ELB) sind grau dargestellt, pVHL orange und ein von HIF-1α abgeleitetes Substratpeptid (Reste 561-575) grün. (B) Detailansicht der HIF-1α-Bindestelle nach Rotation von pVHL um etwa 180° um die vertikale Achse. Der für die Erkennung durch pVHL kritische Hydroxyprolinrest Hyp564 des Peptides ist gelb markiert. Die Seitenketten von Y98 und Y112 sind lila markiert. Beide Aminosäurereste sind an der Bindung des Substratpeptides beteiligt. Mutationen von Y98 und Y112 zu Asparagin (N) führen zu starken Defekten in der Substratbindung und –ubiquitylierung und sind mit einem hohen Risiko von Nierenzell-Karzinomen korreliert (Krankheitstyp 2B). Mutationen zu Histidin (H) verursachen weniger starke Defekte und sind mit einem niedrigen Risiko von Nierenzellkarzinomen korreliert (Krankheitstyp 2A) [5].](/333457/original-1293750029.jpg?t=eyJ3aWR0aCI6MjQ2LCJvYmpfaWQiOjMzMzQ1N30%3D--fe0c6036dcebec9c0add25dacfac188bfe0fc5dd)