Forschungsbericht 2009 - Max-Planck-Institut für Biochemie
Das erste vollständige Proteom
Proteomics und Signaltransduktion (Mann) (Prof. Dr. Matthias Mann)
MPI für Biochemie, Martinsried
Die Verarbeitung der Erbinformation
In jeder Zelle wird Erbinformation, die in der DNA kodiert und gespeichert ist, in Eiweißmoleküle übersetzt. Die daraus entstehenden Proteine unterscheiden sich voneinander durch die Reihenfolge von 20 verschiedenen Aminosäurebausteinen. Proteine gehören zu den aktiven Molekülen der Zelle, während die DNA lediglich als Informationsspeicher dient. Proteine sind zum Beispiel dafür zuständig, der Zelle Struktur zu geben, Nahrung enzymatisch zu spalten und den Informationsfluss der Zelle zu steuern. Während umfassende DNA-Analysen in der Biologie mittlerweile Routine sind und die komplette Erbanlage vieler Arten und Lebewesen bekannt ist, steckt die ebenso wichtige Gesamtanalyse der Proteine eines Organismus vergleichsweise noch in den Kinderschuhen.
Massenspektrometrie als grundlegende Technik der Proteomik
Die Massenspektrometrie ermöglicht es, ein Signal von allen Stoffen zu erhalten, die vorher in das Vakuum des Massenspektrometers überführt und ionisiert wurden. Im Jahre 1988 gelang es John Fenn, Proteine mithilfe des von ihm entwickelten Elektrosprays zu verdampfen und zu ionisieren [1], wofür er 2002 den Nobelpreis in Chemie erhielt. Damit schuf er eine der wichtigsten Voraussetzungen für die Proteomik. Abbildung 1 stellt den Arbeitsgang der modernen Proteomik dar.

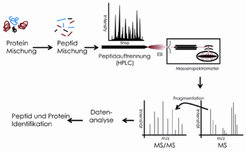
Wir arbeiten intensiv daran, die auf Massenspektrometrie basierte Proteomik leistungsstärker zu machen. So werden beispielsweise Proteine in der Regel mithilfe von Verdauungsenzymen in kurze Proteinbruchstücke verwandelt, die sich dann nachfolgend wesentlich einfacher massenspektrometrisch analysieren lassen. Im vergangenen Jahr entwickelten wurde eine Methode entwickelt, die die Effizienz, mit der Proteine aus Zellen extrahiert und verdaut werden können, stark verbessern konnte (Filter-aided sample preparation, FASP [2]).
Die Massenspektrometer in unserer Abteilung sind sogenannte Orbitraps. Hier umkreisen die Ionen eine Spindel im Hochvakuum, und die gemessene Frequenz ergibt hochaufgelöste und sehr genaue Massenspektren. Im Rahmen von Netzwerken, die von der Europäischen Union gefördert werden, trägt unsere Gruppe zusätzlich zur Weiterentwicklung der Massenspektrometrie bei.
Vergleich biologischer Zustände mit quantitativer Massenspektrometrie

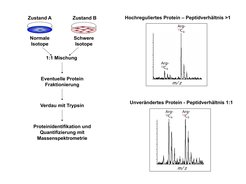
Die Identifikation von Proteinen allein ist nicht sehr aussagekräftig – erst der quantitative Vergleich erlaubt es, biologisches oder medizinisches Wissen zu gewinnen. Unsere Gruppe hat die bisher genaueste Methode der quantitativen Massenspektrometrie entwickelt, das Stable Isotope Labeling by Amino Acids in Cell Culture (SILAC) [3]. Im SILAC-Verfahren wird eine Aminosäure durch eine isotopmarkierte, schwere Variante ersetzt – zum Beispiel das normale Lysin durch 13C6-Lysin. Vermischt man nun Extrakte von Zellen, deren Proteine die herkömmliche Aminosäure enthalten, mit Extrakten von solchen Zellen, deren Proteine die schwere Aminosäure enthalten, kann man die Signale ihrer Peptide im Massenspektrum eindeutig zuordnen. Dadurch können mehrere biologische Zustände in einer einzigen Messung mit größtmöglicher Genauigkeit miteinander verglichen werden. Die SILAC-Methode wird ständig weiterentwickelt und inzwischen weltweit in vielen Labors angewendet (Abb. 2).
Das erste Gesamtproteom
 und dem Verhältnis ihres Vorkommens zwischen haploider und diploider Hefe (x-Achse) dargestellt.")
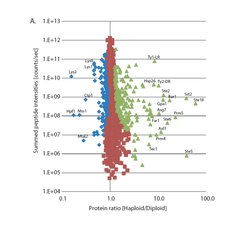
Die Analyse von vielen Proteinen gleichzeitig ist wesentlich schwieriger als die Analyse von DNA oder RNA. Deshalb galt es auch bis vor kurzem als unmöglich, alle Proteine einer Zelle zu messen. Mithilfe der Technologien unserer Abteilung – insbesondere der von Dr. Jürgen Cox entwickelten mathematischen Verfahren, die in die MaxQuant Software einfließen [4], ist es zum ersten Mal gelungen, die Gesamtheit der Proteine eines Lebewesens zu analysieren. Wir verglichen Hefezellen mit einfachem und doppeltem Chromosomensatz (hapliod und diploid) und quantifizierten mehr als 4000 Proteine [5]. Das US Wissenschaftsjournal Science beschrieb dieses erste Gesamtproteom - 12 Jahre nach der DNA-Sequenzierung des ersten Genoms – als einen der zehn wissenschaftlichen Durchbrüche des Jahres (Abb. 3).
Bestimmung von Proteinmodifikationen
Zusätzliche Modifikationen von Proteinen können deren Funktion stark beeinflussen. Eines der wichtigsten Anwendungsgebiete der Proteomik ist die Charakterisierung dieser Modifikationen. Wir haben die SILAC-Methode benutzt, um beispielsweise mehr als 6000 Proteinphosphorylierungen zu quantifizieren, nachdem die Zellen einem Wachstumsfaktor ausgesetzt worden waren [6]. Kürzlich bestimmten wir auch mehr als 20.000 Phosphorylierungsstellen in Krebszellen während des Zellzyklus. Damit konnten wir neue Einblicke in dieses komplexe Regulationssystem gewinnen, das die Teilung der Zelle kontrolliert [7]. Der Zellzyklus spielt unter anderem eine wichtige Rolle bei der Entstehung von Krebs.
Mögliche Anwendungen in der Diagnostik
Ein Teil der Abteilung beschäftigt sich auch mit klinischer Proteomik. Hier geht es darum, aus dem Expressionsmuster der Proteine oder aus ihren Modifikationen Vorhersagen über das Entstehen oder den Ablauf von Krankheiten zu gewinnen. Da ein Einsatz der SILAC-Strategie für menschliche Gewebeproben nicht zugänglich ist, wird eine Variante benutzt, in der mehrere markierte Zelllinien zusammengemischt werden und die deshalb Super-SILAC genannt wird. Mit der Super-SILAC-Methode kann effizient das Proteinmuster von Krebsgeweben oder normalen Geweben quantifiziert werden, und wir hoffen, dass diese Methode in der Zukunft Anwendung finden wird für die Diagnostik vieler Krankheiten.
Ausblick
In den nächsten Jahren wird weiter an der Verbesserung der proteomischen Methoden geforscht. In vielen Bereichen, wie Empfindlichkeit, Durchsatz und Genauigkeit, sind noch mindestens Verbesserungen um das Zehnfache möglich. Ein besonderer Fokus wird zudem auf der Untersuchung anderer Proteinmodifikationen, wie der Acetylierung und Glycosilierung, liegen [8]. Wir erwarten, dass die Technologie zukünftig wesentlich breiter eingesetzt wird und sich zu einem wichtigen Eckpfeiler der modernen Biologie, möglicherweise auch der Medizin, entwickelt.