Forschungsbericht 2018 - Max-Planck-Institute für experimentelle Medizin
Vom Tiermodell zum Patienten: Entwicklung neuer Therapien bei der Charcot-Marie-Tooth Neuropathie (CMT)
Einleitung
Die Charcot-Marie-Tooth Neuropathie (CMT) erfüllt mit einer Häufigkeit von 1:2.500 die Kriterien einer seltenen Erkrankung. Allein in Deutschland finden sich mindestens 30.000 Patienten, weltweit aber sind mehr als 2 Millionen Menschen betroffen, die ohne eine ursächliche Therapie leben müssen. Ursache der häufigsten Unterform CMT1A ist ein genetischer Defekt: die Verdopplung des Gens für das Protein „PMP22“, welche zu einer Fehlfunktion von Gliazellen im peripheren Nervensystem (PNS) führt. Diese „Schwannschen“ Zellen sorgen normalerweise für die Ummantelung von Nervenzellfortsätzen (Axonen) mit einer isolierenden, fettreichen Schicht, dem Myelin. Sie gewährleistet eine schnelle Weiterleitung von elektrischen Impulsen und ermöglicht so eine schnelle Reaktion auf einen Reiz, zum Beispiel eine Muskelkontraktion. CMT1A-Patienten leiden an einer langsam fortschreitenden Nervenschädigung in den Beinen und Armen, die zu Muskelschwäche und -schwund führt. Bereits im Kindesalter können Fußdeformitäten, Gehbehinderungen und Gefühlsstörungen die Folge sein. Bisher wurden zwar einige wenige Therapien in Tiermodellen erprobt, aber noch nie erfolgreich auf menschliche Patienten übertragen.
Lipide in der Nahrung verbessern die Pathologie der Erkrankung
Um in den ersten drei Wochen nach der Geburt das lipidreiche Myelin bilden zu können, werden bei normalem Entwicklungsverlauf vermehrt die am Fettsäurestoffwechsel beteiligten Gene „angeschaltet“. Bereits in vorangegangenen Arbeiten konnten wir zeigen, dass in PMP22-transgenen Ratten, einem von uns entwickelten Tiermodell für die CMT1A-Erkrankung („CMT1A-Ratte“), die Aktivierung von Lipidgenen ausbleibt. Da das Myelin bei CMT niemals richtig aufgebaut wird, könnte die fehlende Lipidsynthese für die gestörte Myelinisierung bei CMT-Patienten mitverantwortlich sein [1, 2].
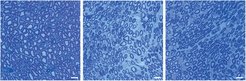
Im Rahmen einer aktuellen Studie des vom Bundesministerium für Bildung und Forschung (BMBF) geförderten Forschungsnetzwerkes „CMT-NET“ haben wir uns mit der Frage beschäftigt, ob diese beeinträchtigte Fettsäuresynthese behandelt werden kann. Dabei untersuchten wir, ob von außen (exogen) mit der Nahrung zugeführte Lipide die Myelinisierung und die Muskelkraft in CMT1A-Ratten verbessern. Nach ermutigenden Vorversuchen, die uns zeigten, dass exogen zugefügte Lipide in vitro (in Zellkultur) und in vivo (im lebendigen Versuchstier) von Schwann´schen Zellen in das Myelin eingebaut werden, wurde eine proof-of-principal (dem Grundsatzbeweis dienende) tierexperimentelle Studie in CMT1A-Ratten durchgeführt. Die Tiere bzw. die säugenden Mütter (CMT1A-Ratten sowie gesunde Kontrollen) wurden ab dem 2. Tag nach der Geburt über 16 Wochen mit dem lipidreichen Nahrungsergänzungsmittel Lecithin, einer aus Soja oder Eigelb gewonnenen Mischung aus Phospholipiden, gefüttert. Erfreulicherweise zeigten die behandelten CMT1A-Ratten am Ende der Studie eine mit den gesunden Kontrollratten vergleichbare deutlich gesteigerte Muskelkraft, verbunden mit einer entsprechend erhöhten Anzahl myelinisierter Axone ([3], Abb. 1).
 sind in CMT1A-Ratten (mittleres Bild) im Nervenquerschnitt weniger mit Myelin ummantelte Nervenfasern zu sehen, erkennbar als blaue Ringe. Eine Therapie von CMT1A Ratten mit Lecithin (rechtes Bild) erhöht die Anzahl myelinisierter Fasern.")
Weitere Experimente belegten, dass die Zusammensetzung des Myelins (Verhältnis von Lipiden zu Proteinen) in CMT1A-Ratten nach der Therapie verbessert und die Lipidtherapie nicht von einem bestimmten Alters- und Entwicklungsfenster der Ratten abhängig ist. CMT1A-Tiere, die früh in der Entwicklung während der aktiven Myelinisierung kurzzeitig für 3 Wochen behandelt wurden, zeigten bereits am Therapieende verbesserte motorische Fähigkeiten verbunden mit einer erhöhten Anzahl myelinisierter Fasern. Auch ein Therapiebeginn in erwachsenen Tieren führte zu einer deutlichen Verbesserung der Krankheitssymptomatik im CMT1A-Rattenmodell. Die Wirksamkeit der Lecithin-Therapie unabhängig vom Behandlungsbeginn im Rattenmodell zusammen mit der bereits belegten guten Verträglichkeit von Lecithin in Menschen macht den hier identifizierten Behandlungsansatz zu einem vielversprechenden Therapeutikum für CMT1A-Patienten, der womöglich auch auf andere, ähnliche Neuropathien mit Myelinisierungs-Störungen übertragen werden könnte. Eine entsprechende klinische Studie mit CMT1A-Patienten befindet sich derzeit in Planung.
Ausblick
Bereits in früheren präklinischen Studien konnten wir für CMT1A-Ratten zeigen, dass sich durch die pharmakologische Blockade des Progesteron-Rezeptors die krankheitsverursachende Überexpression von Pmp22-Gentranskripten korrigieren und damit der Verlauf der Erkrankung behandeln lässt [4, 5]. Leider sind Progesteron-Antagonisten für die langfristige Anwendung nicht geeignet. In Zusammenarbeit mit einem industriellen Partner erfolgte deshalb eine Weiterentwicklung des Konzepts der pharmakologischen Transkriptionskontrolle: PXT3003 ist ein Kombinationspräparat aus den bereits zugelassenen und damit für den Patienten sicheren Arzneistoffe Baclofen, Naltrexon und Sorbitol. Diese drei Komponenten wirken synergistisch auf die Pmp22-Überexpression, aber auch auf weitere krankheitsrelevante Signalwege, ohne dass diese Interaktion ganz genau verstanden ist. Aufbauend auf die vielversprechenden präklinischen Daten der mit PXT3003 behandelten CMT1A-Ratten wurde eine explorative Phase 2 klinische Studie durchgeführt, an die sich 2016 eine doppelverblindete, internationale und multizentrische klinische Zulassungsstudie der Phase 3 (RCT) mit ca. 300 CMT1A-Patienten anschloss. Diese konnte im Oktober 2018 mit überraschend positiven Ergebnissen die Wirksamkeit von PXT3003 bekanntgeben.
PXT3003 ist damit das erste Medikament gegen eine erbliche Neuropathie, das erfolgreich aus der präklinischen Forschung heraus entwickelt wurde. Es wird voraussichtlich 2020 in USA und Europa erhältlich sein. Auf www.cmt-net.de, einer Plattform, die von Göttingen aus koordiniert wird, können sich Patienten, Wissenschaftler und Ärzte über die Fortschritte bei der Erforschung dieser häufig nicht erkannten Erkrankung informieren.
Literaturhinweise
British Medical Bulletin 102, 89-113 (2012)
Erratum in: Annals of Neurology 61(3): 282 (2007)